We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
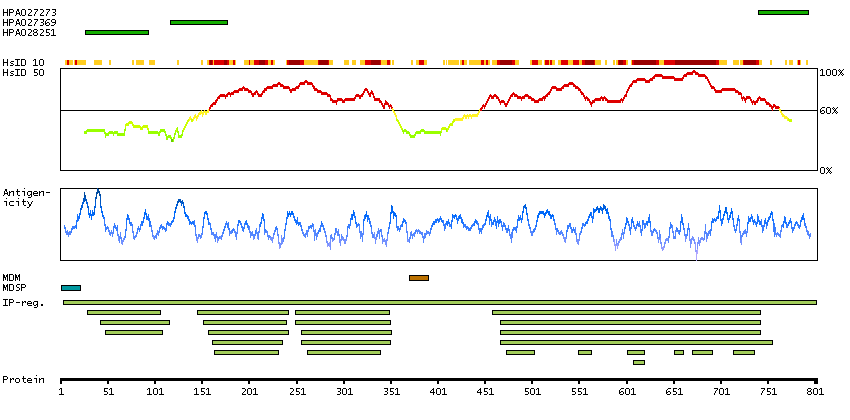
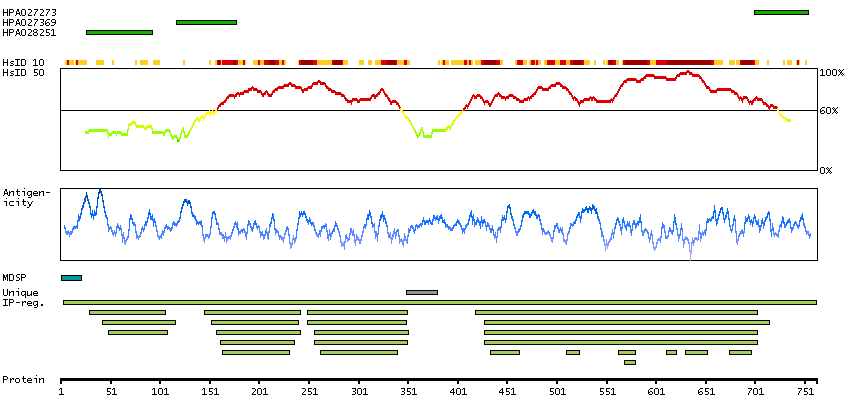
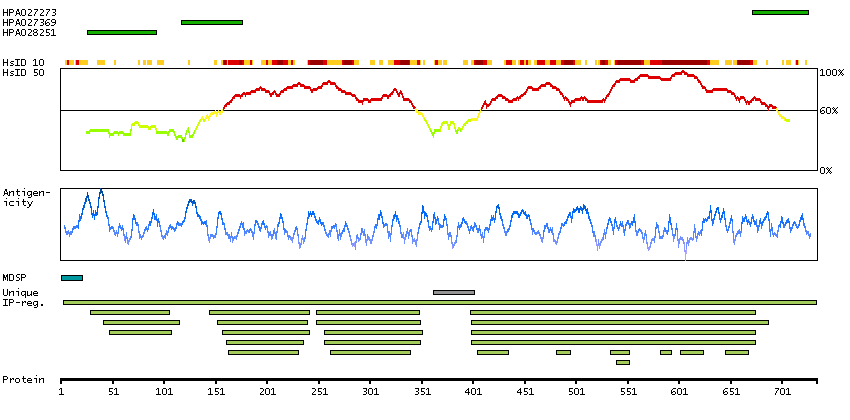
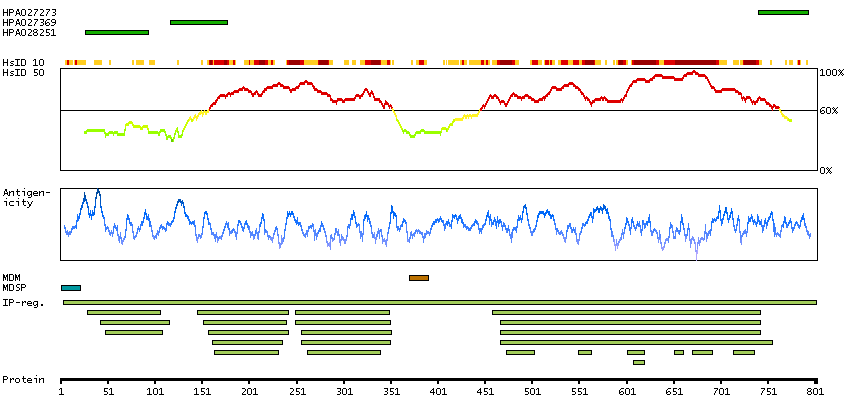
The protein encoded by this gene is a member of the fibroblast growth factor receptor family, where amino acid sequence is highly conserved between members and throughout evolution. FGFR family members differ from one another in their ligand affinities and tissue distribution. A full-length representative protein would consist of an extracellular region, composed of three immunoglobulin-like domains, a single hydrophobic membrane-spanning segment and a cytoplasmic tyrosine kinase domain. The extracellular portion of the protein interacts with fibroblast growth factors, setting in motion a cascade of downstream signals, ultimately influencing mitogenesis and differentiation. The genomic organization of this gene, compared to members 1-3, encompasses 18 exons rather than 19 or 20. Although alternative splicing has been observed, there is no evidence that the C-terminal half of the IgIII domain of this protein varies between three alternate forms, as indicated for members 1-3. This particular family member preferentially binds acidic fibroblast growth factor and, although its specific function is unknown, it is overexpressed in gynecological tumor samples, suggesting a role in breast and ovarian tumorigenesis. [provided by RefSeq, Jul 2008]