We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
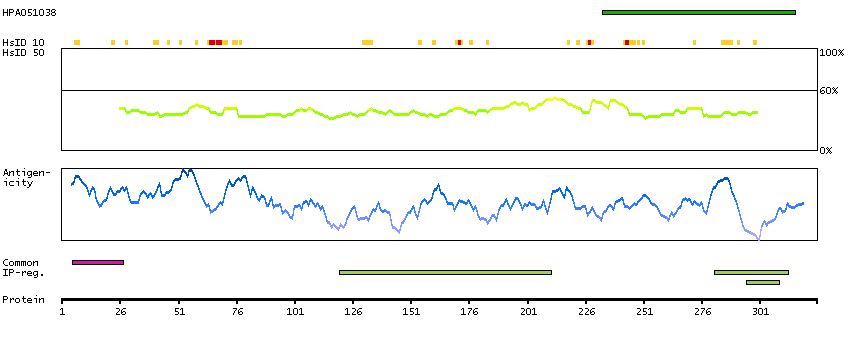
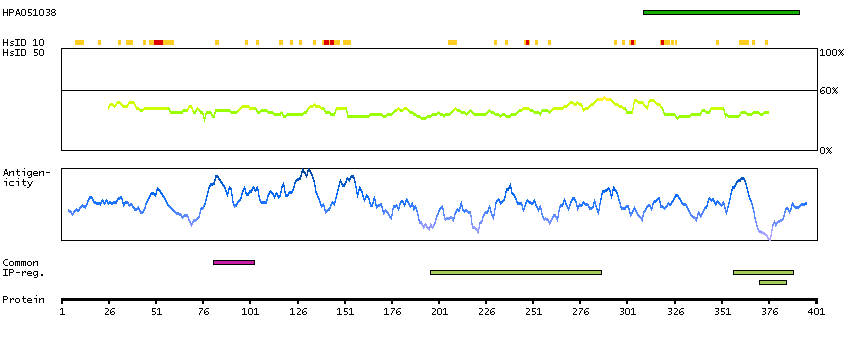
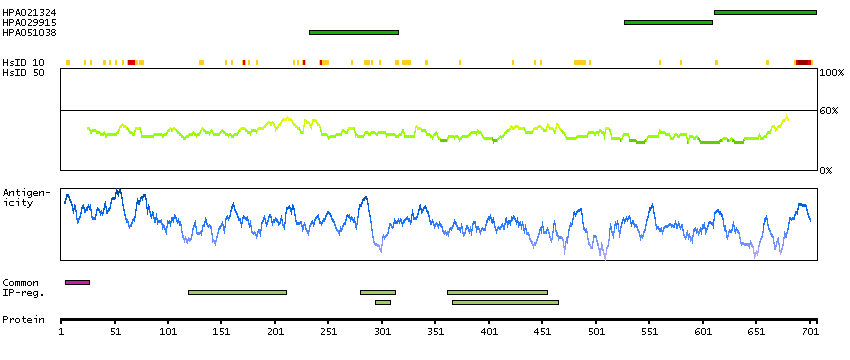
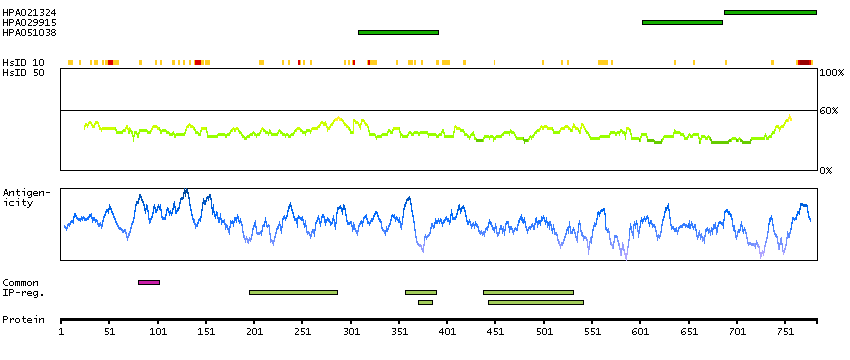
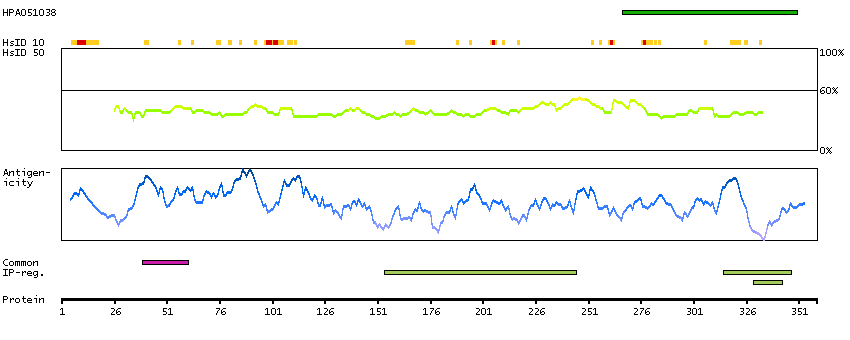
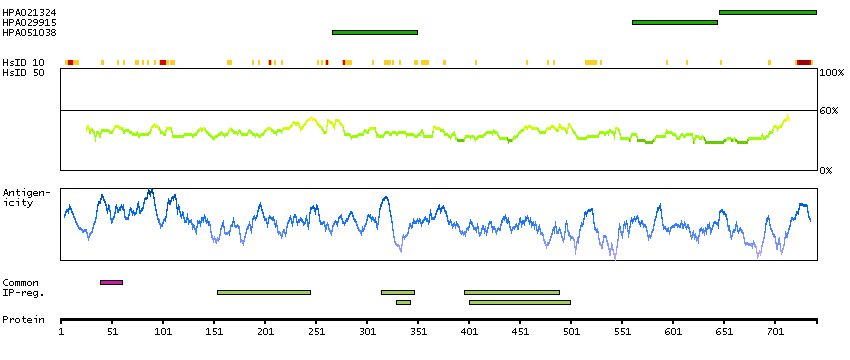
This is a paternally expressed imprinted gene that is thought to have been derived from the Ty3/Gypsy family of retrotransposons. It contains two overlapping open reading frames, RF1 and RF2, and expresses two proteins: a shorter, gag-like protein (with a CCHC-type zinc finger domain) from RF1; and a longer, gag/pol-like fusion protein (with an additional aspartic protease motif) from RF1/RF2 by -1 translational frameshifting (-1 FS). While -1 FS has been observed in RNA viruses and transposons in both prokaryotes and eukaryotes, this gene represents the first example of -1 FS in a eukaryotic cellular gene. This gene is highly conserved across mammalian species and retains the heptanucleotide (GGGAAAC) and pseudoknot elements required for -1 FS. It is expressed in adult and embryonic tissues (most notably in placenta) and reported to have a role in cell proliferation, differentiation and apoptosis. Overexpression of this gene has been associated with several malignancies, such as hepatocellular carcinoma and B-cell lymphocytic leukemia. Knockout mice lacking this gene showed early embryonic lethality with placental defects, indicating the importance of this gene in embryonic development. Additional isoforms resulting from alternatively spliced transcript variants, and use of upstream non-AUG (CUG) start codon have been reported for this gene. [provided by RefSeq, Oct 2014]