We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
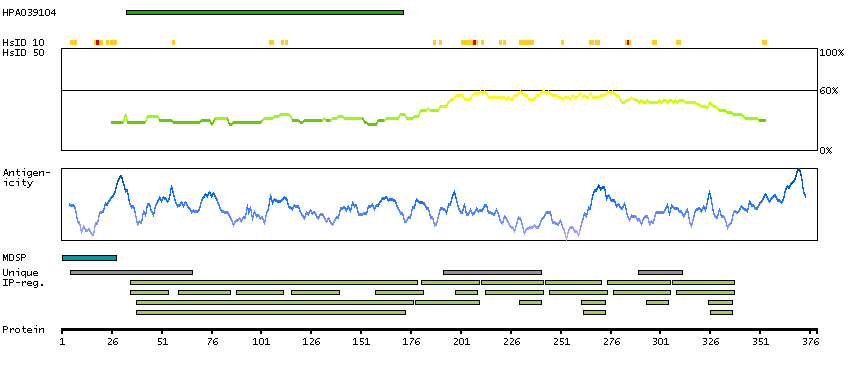
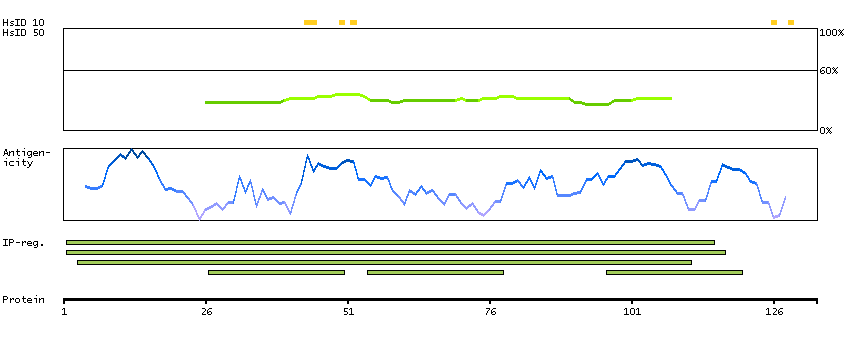
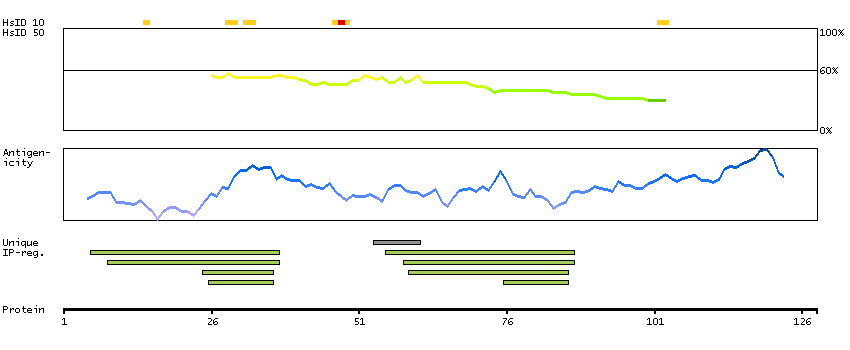
The protein encoded by this gene functions to inhibit WNT proteins, which are extracellular signaling molecules that play a role in embryonic development. This protein contains a WNT inhibitory factor (WIF) domain and five epidermal growth factor (EGF)-like domains, and is thought to be involved in mesoderm segmentation. This gene functions as a tumor suppressor gene, and has been found to be epigenetically silenced in various cancers. [provided by RefSeq, Jun 2010]
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)