We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
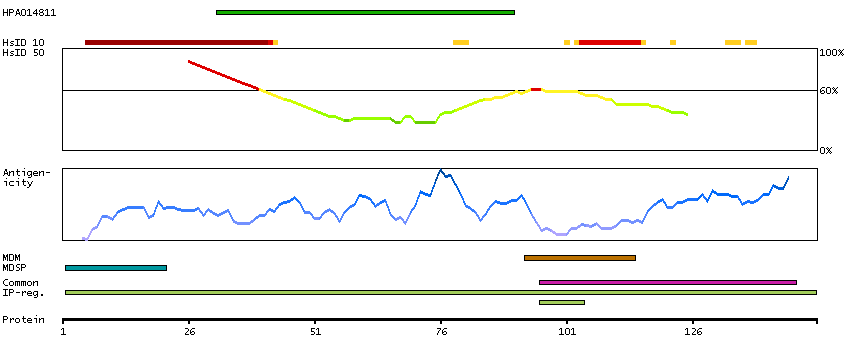
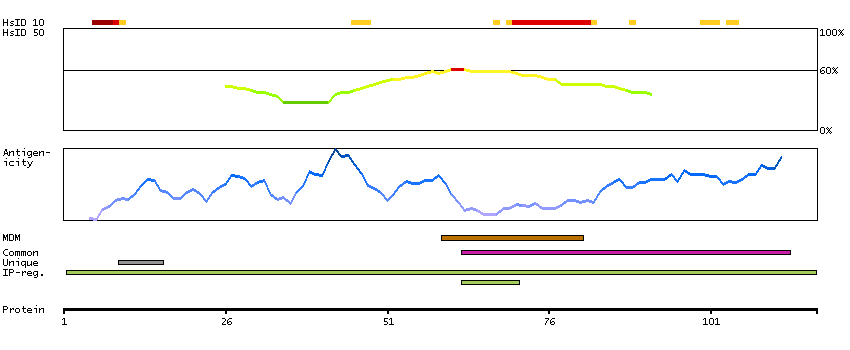
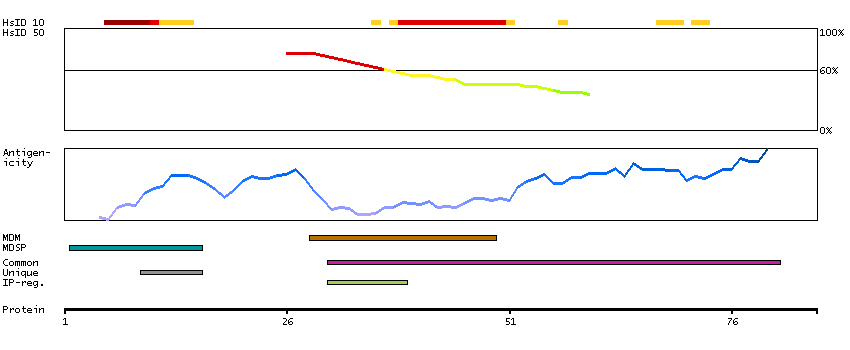
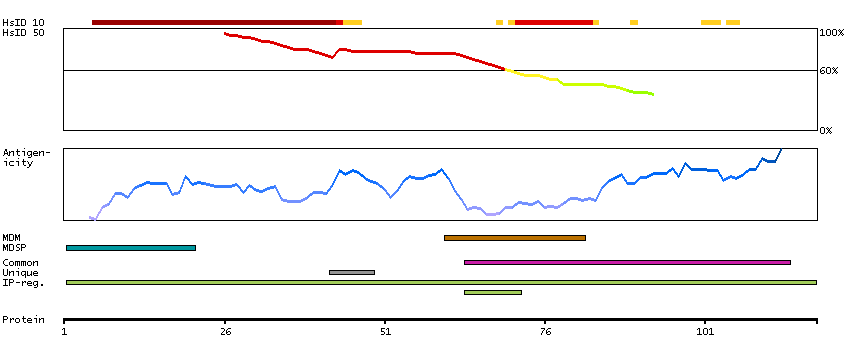
Glycophorins A (GYPA) and B (GYPB) are major sialoglycoproteins of the human erythrocyte membrane which bear the antigenic determinants for the MN and Ss blood groups. In addition to the M or N and S or s antigens that commonly occur in all populations, about 40 related variant phenotypes have been identified. These variants include all the variants of the Miltenberger complex and several isoforms of Sta, as well as Dantu, Sat, He, Mg, and deletion variants Ena, S-s-U- and Mk. Most of the variants are the result of gene recombinations between GYPA and GYPB. [provided by RefSeq, Jul 2008]
GO:0005887 [integral component of plasma membrane] GO:0007016 [cytoskeletal anchoring at plasma membrane] GO:0009897 [external side of plasma membrane] GO:0016021 [integral component of membrane] GO:0042803 [protein homodimerization activity] GO:0047484 [regulation of response to osmotic stress]