We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Distinct positivity in plasma, extracellular matrix and proximal renal tubules.
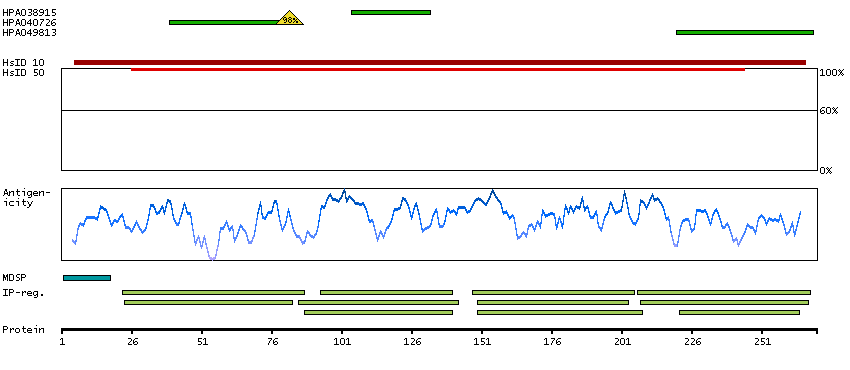
DATA RELIABILITY
Data reliability description
Caution, targets protein from more than one gene. Secreted protein, tissue location of RNA and protein might differ and correlation is complex. Antibody staining in cells/structures not annotated, view images.