|
METHODS
|
Immunoproteomics
Introduction
Mass spectrometry (MS)-based proteomics has in recent years become a method of choice to study proteins, as MS is a great technique for protein identification (Aebersold & Mann, 2003) and offers high precision, good linear range, and good sensitivity. However, so far Enzyme Linked Immunosorbent Assays (ELISAs) and similar antibody-based methods excel MS methods when it comes to sensitive detection of low levels of a protein in complex matrices, whereas MS enables unbiased approaches and can provide unsurpassed specificity. In recent years, more targeted MS technologies have emerged to overcome limitations originating from the inherited problems associated with data dependent scanning during sampling (Gillette & Carr, 2013), resulting in decreased time needed for analysis. A limiting factor when using MS methods is the large differences in concentration range between highly abundant proteins in very complex matrices, e.g. serum or plasma, and low abundant proteins. The fact that the dynamic range in proteomes often range several orders of magnitude between high and low abundant proteins (Surinova et al., 2011) often makes it necessary to use protein depletion of the most abundant proteins and/or elaborate fractions before running the MS analysis. The dynamic range problem has prompted several investigators to introduce a protein or peptide capture step using specific antibodies to allow for immuno-affinity enrichment prior the mass spectrometry analysis (Berna et al., 2008). By enriching target proteins or peptides using antibodies in such an approach, the complexity of the sample is drastically reduced and the protein or peptide derived from the antibody reagent can successfully be identified by MS. This decreases the demand on long and time-consuming liquid chromatography gradients. Hence, the combination of immuno-based methods with mass spectrometry detection has become an indispensable tool in the emerging field of quantitative proteomics.
Technology
Assay design
An attractive strategy for quantitative proteomics using immuno-enrichment is to include stable isotope standards in the assay format. Stable isotope labeled peptides (Barr et al., 1996; Gerber, Rush, Stemman, Kirschner, & Gygi, 2003), proteins (Brun et al., 2007; Singh, Springer, Steen, Kirschner, & Steen, 2009), or protein fragments (Zeiler, Straube, Lundberg, Uhlen, & Mann, 2012) are added to the sample and used for absolute protein quantification by MS. The approach of stable isotope labeling of the reference standards is either accomplished by chemical addition of labeled reagents, enzymatic isotope labeling, or metabolic labeling. The quantitative information about the endogenous target can be acquired from MS data by comparing ratios between the endogenous peptide and the heavy labeled spiked in peptide. By combining the isotope standards with immuno-enrichment using antibodies, absolute quantitative data can be gathered from complex biological samples in an efficient way. However, the major disadvantage with the immuno-affinity proteomics strategy is the limited availability of suitable antibodies that efficiently recognize peptides from the corresponding protein targets. The enrichment of peptides usually requires generation of custom antibodies for each target peptide and this very time-consuming process makes high-throughput efforts very difficult to pursue. Most efforts so far have been aimed towards generating monoclonal antibodies against specific selected peptides to be appropriate for MS-detection, which is a laborious and costly exercise.
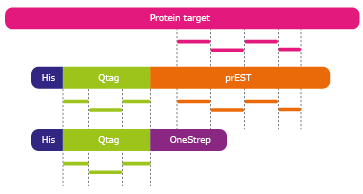
A semi-automated method called "Immuno-SILAC", where SILAC stands for Stable Isotope Labeling by Amino acids in Cell culture, has been developed for multiplex peptide enrichment using SILAC-standards and polyclonal antibodies immobilized on protein A-coated magnetic beads. The Immuno-SILAC method is based on antibodies from the Human Protein Atlas (HPA) initiative in combination with pre-digestion addition of heavy isotope labeled recombinant protein fragment standards. The different fragments used in Immuno-SILAC are schematically described in Figure 1.
Figure 1. The different fragments used in immuno-SILAC. Absolute protein quantification using Quantitative PrESTs (orange) as internal standard. Peptides originating from the Q-tag (green), are used to quantify
each PrEST against an ultra purified Qtag protein standard (third fragment from top). The PrEST can thereafter be used as an internal standard in unknown samples to quantify its corresponding endogenous protein
(pink) in LC-MS.
The HPA project produce polyclonal antibodies that recognize linear epitopes and the project have a large library of recombinant protein fragments called Protein Epitope Signature Tags (PrESTs),
which have been shown to be a valuable source as isotopic internal standards (Zeiler et al., 2012). First the isotopic standards, called quantitative PrESTs (QPrESTs),
are produced as a fusion protein together with an Q-tag in Escherichia coli and heavy isotope-labeled versions (15N and 13C) of the amino acids arginine and lysine are incorporated in the
protein sequence. Each QPrEST is then accurately quantified by MS using a non labeled absolutely quantified Q-tag protein. Thereafter, known amounts of QPrEST can be spiked into unknown biological samples as standards.
This is preferably done before enzyme digestion to allow the heavy protein standards to undergo the same processing as the endogenous proteins throughout sample preparation, compensating for miscleavages or otherwise incomplete digestion. The mix of standard and sample is enzymatically
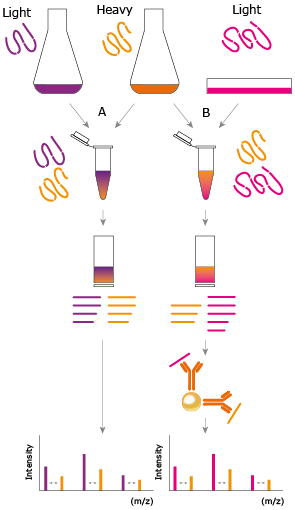
cleaved and after affinity enrichment using polyclonal antibodies, the SILAC ratios may be used to determine absolute protein quantities of endogenous proteins, Figure 2.
Figure 2. General principle of immuno-SILAC. Representation of the Immuno-SILAC workflow. Highly purified and accurately quantified isotopic heavy labeled PrESTs (orange) are spiked into cell lysates containing ultra purified standard (purple) or unknown sample (pink) prior trypsin digestion, thereby minimizing the risk of differences arising between samples and standards during sample preparation. Antibodies coupled to magnetic solid-phase support enrich target peptides from the digested sample and the endogenous protein concentration is calculated from the ratio of heavy to light peptides detected by MS.
Specific examples
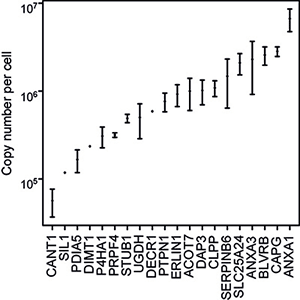
A quantitative multiplex analysis using 41 antibodies against heavy labeled PrESTs was performed in a human HeLa cell lysate (Edfors et al., 2014). Here, twentytwo antibodies, corresponding to 20 protein targets, successfully enriched peptides that later were used for quantification after MS analysis. Among the 20 identified proteins, copy numbers ranged from 6.6 million copies per cell for annexin 1 (ANXA1) down to the 57000 copies per cell determined for calcium activated nucleotidase (CANT1), Figure 3. For immuno-SILAC, 15 µg of a digested HeLa sample was used in the peptide enrichment prior to MS analysis and only a 15-minute HPLC gradient is needed for sufficient peptide separation.
Figure 3. Immuno-SILAC quantification of 20 target proteins (x-axis) as copy number per cell (y-axis). The analysis was performed in triplicates and median values were plotted with error bars showing the standard deviation.
References and Links
- An article about mass spectrometry-based proteomics:
Aebersold, R., & Mann, M. (2003). Mass spectrometry-based proteomics. Nature, 422(6928), 198-207. doi:10.1038/nature01511.
DOI:10.1038/nature01511. PubMed: 12634793
- An article describing quantification of peptides using isotope dilution for reference concentrations of proteins:
Barr, J. R., Maggio, V. L., Patterson, D. G., Cooper, G. R., Henderson, L. O., Turner, W. E., ... Sampson, E. J. (1996). Isotope dilution--mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clinical Chemistry, 42(10), 1676-82.
PubMed: 8855153
- An article describin immuno-precipitation prior trypsin digestion and mass spectrometry:
Berna, M., Ott, L., Engle, S., Watson, D., Solter, P., & Ackermann, B. (2008). Quantification of NTproBNP in rat serum using immunoprecipitation and LC/MS/MS: a biomarker of drug-induced cardiac hypertrophy. Analytical Chemistry, 80(3), 561-6.
DOI:10.1021/ac702311m. PubMed: 18179251
- An article about mass spectrometry based quantification and isotope-labeled protein standards:
Brun, V., Dupuis, A., Adrait, A., Marcellin, M., Thomas, D., Court, M., ... Garin, J. (2007). Isotope-labeled protein standards: toward absolute quantitative proteomics. Molecular & Cellular Proteomics: MCP, 6(12), 2139-49.
DOI:10.1074/mcp.M700163-MCP200. PubMed: 17848587
- An article about the immuno-SILAC technology using polyclonal antibodies from the Human Protein Atlas:
Edfors, F., Boström, T., Forsström, B., Zeiler, M., Johansson, H., Lundberg, E., ... Uhlen, M. (2014). Immuno-proteomics using polyclonal antibodies and stable isotope labeled affinity-purified recombinant proteins. Molecular & Cellular Proteomics: MCP, 13(6), 1611-1624.
DOI:10.1074/mcp.M113.034140. PubMed: 24722731
- An article absolute quantification of proteins from cell lysates by mass spectrometry:
Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W., & Gygi, S. P. (2003). Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proceedings of the National Academy of Sciences of the United States of America, 100(12), 6940-5.
DOI:10.1073/pnas.0832254100. PubMed: 12771378
- An article about quantitative analysis of peptides and targeted mass spectrometry:
Gillette, M. A., & Carr, S. A. (2013). Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nature Methods, 10(1), 28-34.
DOI:10.1038/nmeth.2309. PubMed: 23269374
- An article absolute protein quantification of proteins using full-length expressed stable isotope-labeled proteins as standards:
Singh, S., Springer, M., Steen, J., Kirschner, M. W., & Steen, H. (2009). FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. Journal of Proteome Research, 8(5), 2201-10.
DOI:10.1021/pr800654s. PubMed: 19344176
- An article about protein biomarkers in plasma:
Surinova, S., Schiess, R., Hüttenhain, R., Cerciello, F., Wollscheid, B., & Aebersold, R. (2011). On the development of plasma protein biomarkers. Journal of Proteome Research, 10(1), 5-16.
DOI:10.1021/pr1008515. PubMed: 21142170
- An article about Protein Epitope Signature Tags (PrESTs) as reagents for proteomics:
Zeiler, M., Straube, W. L., Lundberg, E., Uhlen, M., & Mann, M. (2012). A Protein Epitope Signature Tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Molecular & Cellular Proteomics: MCP, 11(3), O111.009613.
DOI:10.1074/mcp.O111.009613. PubMed: 21964433
|
|