We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
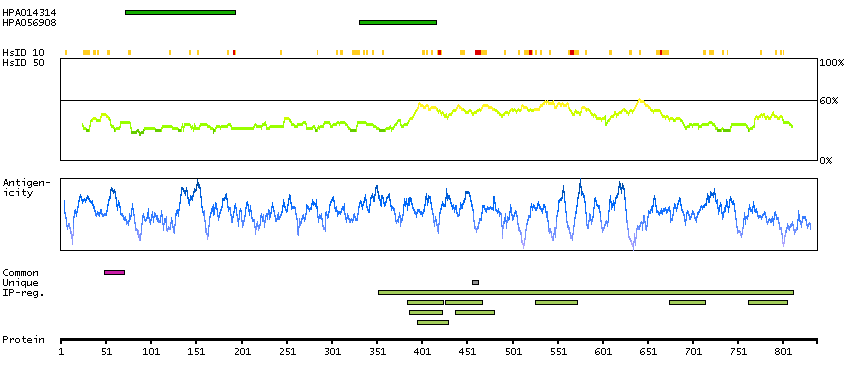
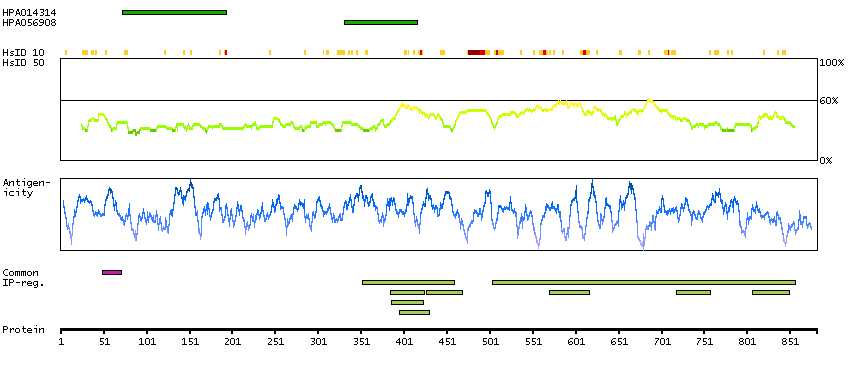
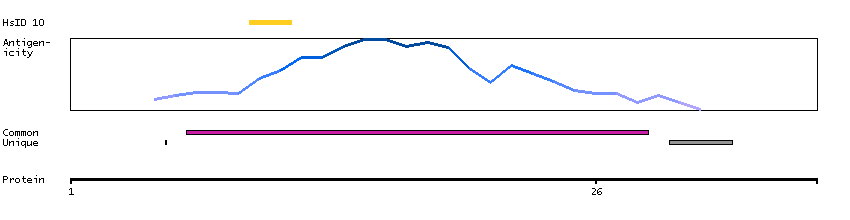
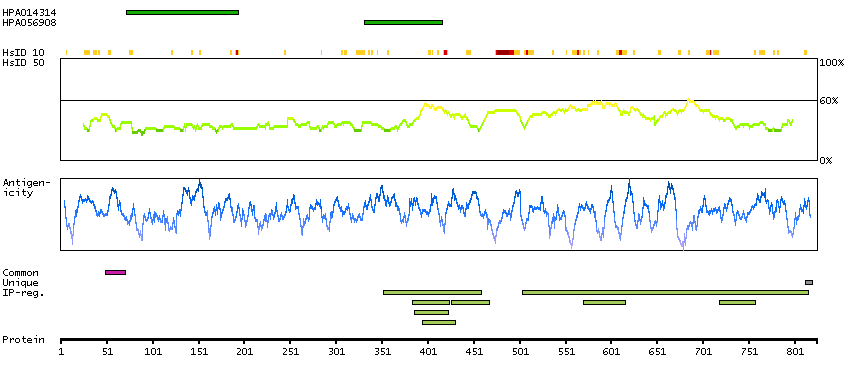
This gene encodes a member of the arm-repeat (armadillo) and plakophilin gene families. Plakophilin proteins contain numerous armadillo repeats, localize to cell desmosomes and nuclei, and participate in linking cadherins to intermediate filaments in the cytoskeleton. This gene product may regulate the signaling activity of beta-catenin. Two alternately spliced transcripts encoding two protein isoforms have been identified. A processed pseudogene with high similarity to this locus has been mapped to chromosome 12p13. [provided by RefSeq, Jul 2008]
SCAMPI predicted membrane proteins Predicted intracellular proteins Plasma proteins Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0002159 [desmosome assembly] GO:0003674 [molecular_function] GO:0005080 [protein kinase C binding] GO:0005488 [binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005882 [intermediate filament] GO:0005886 [plasma membrane] GO:0005911 [cell-cell junction] GO:0005912 [adherens junction] GO:0007507 [heart development] GO:0008285 [negative regulation of cell proliferation] GO:0010765 [positive regulation of sodium ion transport] GO:0014704 [intercalated disc] GO:0016021 [integral component of membrane] GO:0016264 [gap junction assembly] GO:0016337 [single organismal cell-cell adhesion] GO:0017080 [sodium channel regulator activity] GO:0019215 [intermediate filament binding] GO:0030054 [cell junction] GO:0030057 [desmosome] GO:0030336 [negative regulation of cell migration] GO:0032947 [protein complex scaffold] GO:0034334 [adherens junction maintenance] GO:0044325 [ion channel binding] GO:0045110 [intermediate filament bundle assembly] GO:0045294 [alpha-catenin binding] GO:0048496 [maintenance of organ identity] GO:0055010 [ventricular cardiac muscle tissue morphogenesis] GO:0055088 [lipid homeostasis] GO:0086001 [cardiac muscle cell action potential] GO:0086002 [cardiac muscle cell action potential involved in contraction] GO:0086005 [ventricular cardiac muscle cell action potential] GO:0086019 [cell-cell signaling involved in cardiac conduction] GO:0086064 [cell communication by electrical coupling involved in cardiac conduction] GO:0086073 [bundle of His cell-Purkinje myocyte adhesion involved in cell communication] GO:0086083 [cell adhesive protein binding involved in bundle of His cell-Purkinje myocyte communication] GO:0086091 [regulation of heart rate by cardiac conduction] GO:0090002 [establishment of protein localization to plasma membrane] GO:0098911 [regulation of ventricular cardiac muscle cell action potential] GO:2000810 [regulation of bicellular tight junction assembly]