We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
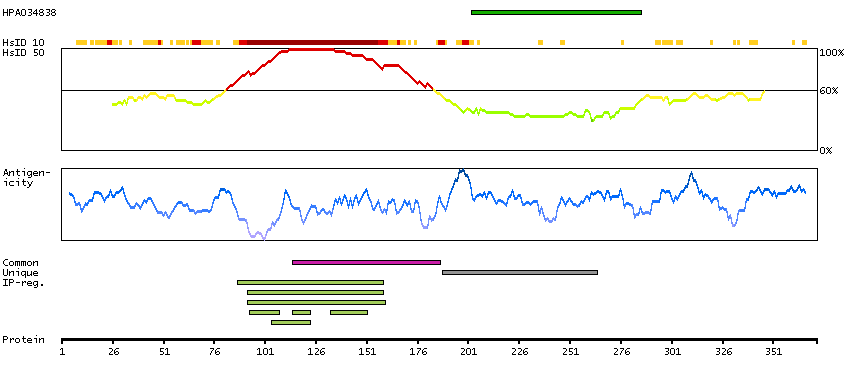
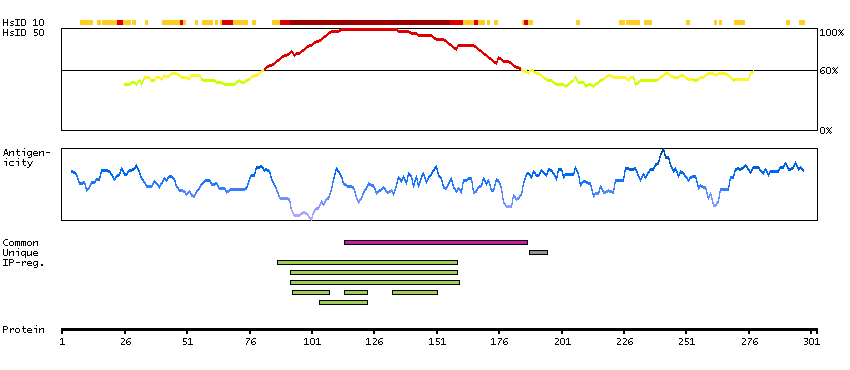
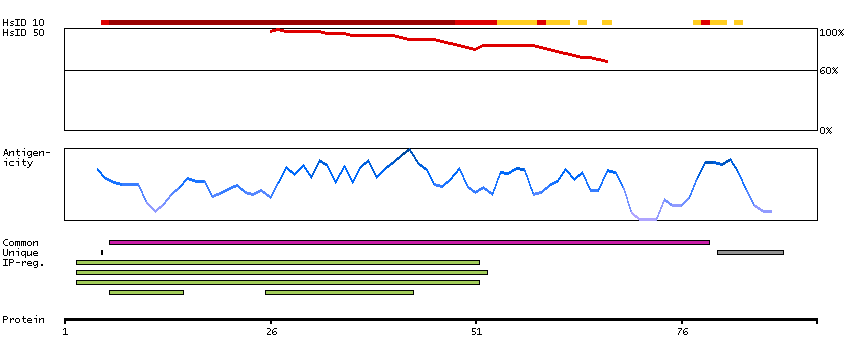
P16989 [Direct mapping] Y-box-binding protein 3 A0A024RAQ1 [Target identity:100%; Query identity:100%] Cold shock domain protein A, isoform CRA_a
Show all
SPOCTOPUS predicted secreted proteins Predicted intracellular proteins Transcription factors beta-Barrel DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0001227 [transcriptional repressor activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0001701 [in utero embryonic development] GO:0003676 [nucleic acid binding] GO:0003677 [DNA binding] GO:0003690 [double-stranded DNA binding] GO:0003697 [single-stranded DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003714 [transcription corepressor activity] GO:0003730 [mRNA 3'-UTR binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005737 [cytoplasm] GO:0005844 [polysome] GO:0005923 [bicellular tight junction] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0007283 [spermatogenesis] GO:0008584 [male gonad development] GO:0009409 [response to cold] GO:0009566 [fertilization] GO:0017048 [Rho GTPase binding] GO:0030529 [ribonucleoprotein complex] GO:0043066 [negative regulation of apoptotic process] GO:0044822 [poly(A) RNA binding] GO:0046622 [positive regulation of organ growth] GO:0048471 [perinuclear region of cytoplasm] GO:0048642 [negative regulation of skeletal muscle tissue development] GO:0060546 [negative regulation of necroptotic process] GO:0070935 [3'-UTR-mediated mRNA stabilization] GO:0071356 [cellular response to tumor necrosis factor] GO:0071474 [cellular hyperosmotic response] GO:1902219 [negative regulation of intrinsic apoptotic signaling pathway in response to osmotic stress] GO:2000767 [positive regulation of cytoplasmic translation]
P16989 [Direct mapping] Y-box-binding protein 3 A0A024RAV4 [Target identity:100%; Query identity:100%] Cold shock domain protein A, isoform CRA_b
Show all
SPOCTOPUS predicted secreted proteins Predicted intracellular proteins Transcription factors beta-Barrel DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003676 [nucleic acid binding] GO:0003677 [DNA binding] GO:0003690 [double-stranded DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003714 [transcription corepressor activity] GO:0003730 [mRNA 3'-UTR binding] GO:0005634 [nucleus] GO:0005737 [cytoplasm] GO:0005923 [bicellular tight junction] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0009409 [response to cold] GO:0017048 [Rho GTPase binding] GO:0044822 [poly(A) RNA binding] GO:0048471 [perinuclear region of cytoplasm] GO:0060546 [negative regulation of necroptotic process] GO:0070935 [3'-UTR-mediated mRNA stabilization] GO:0071356 [cellular response to tumor necrosis factor] GO:0071474 [cellular hyperosmotic response] GO:1902219 [negative regulation of intrinsic apoptotic signaling pathway in response to osmotic stress] GO:2000767 [positive regulation of cytoplasmic translation]