We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
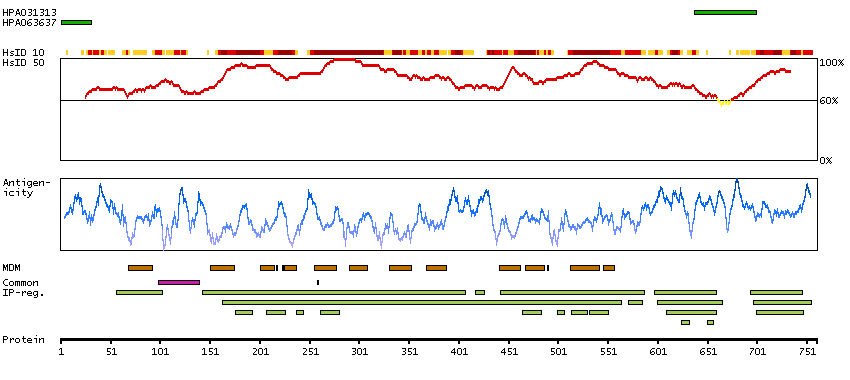
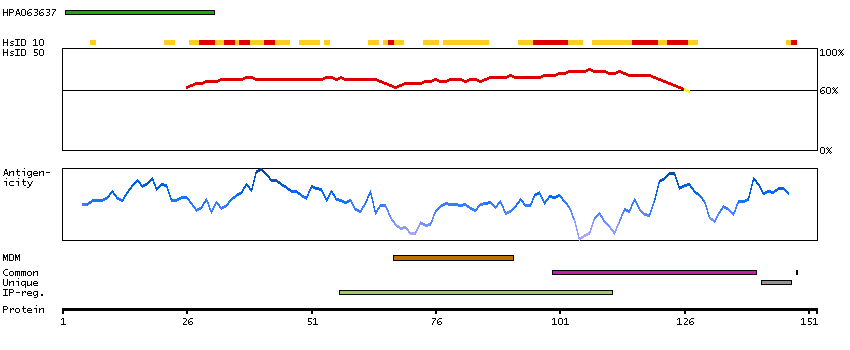
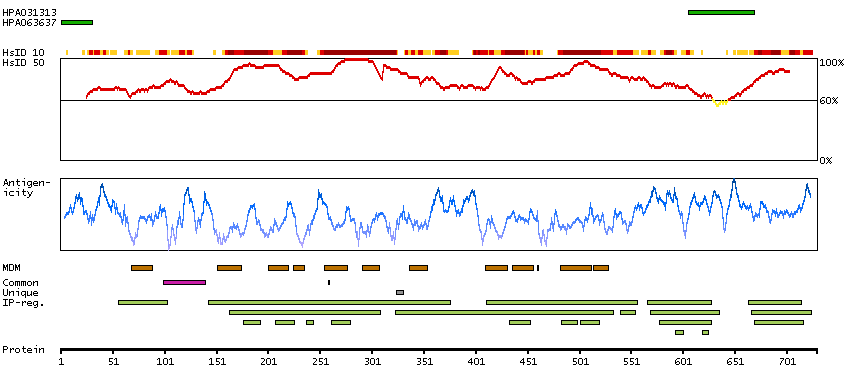
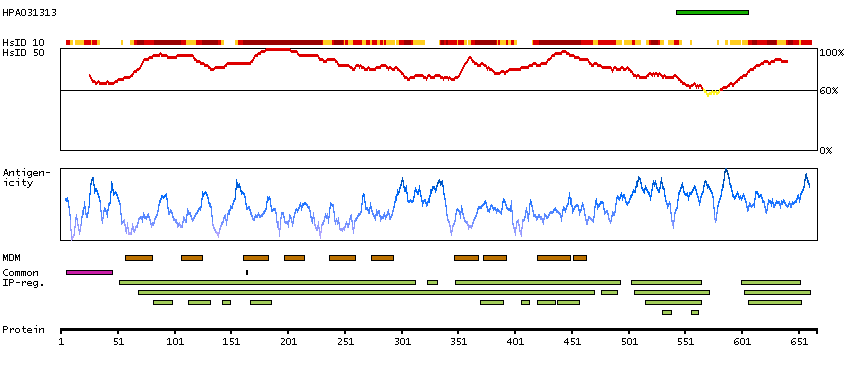
The CLCN family of voltage-dependent chloride channel genes comprises nine members (CLCN1-7, Ka and Kb) which demonstrate quite diverse functional characteristics while sharing significant sequence homology. Chloride channel 4 has an evolutionary conserved CpG island and is conserved in both mouse and hamster. This gene is mapped in close proximity to APXL (Apical protein Xenopus laevis-like) and OA1 (Ocular albinism type I), which are both located on the human X chromosome at band p22.3. The physiological role of chloride channel 4 remains unknown but may contribute to the pathogenesis of neuronal disorders. Alternate splicing results in two transcript variants that encode different proteins. [provided by RefSeq, Mar 2012]