We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
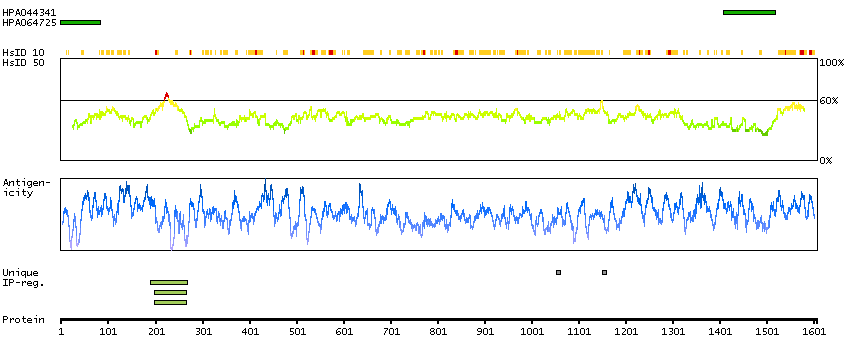
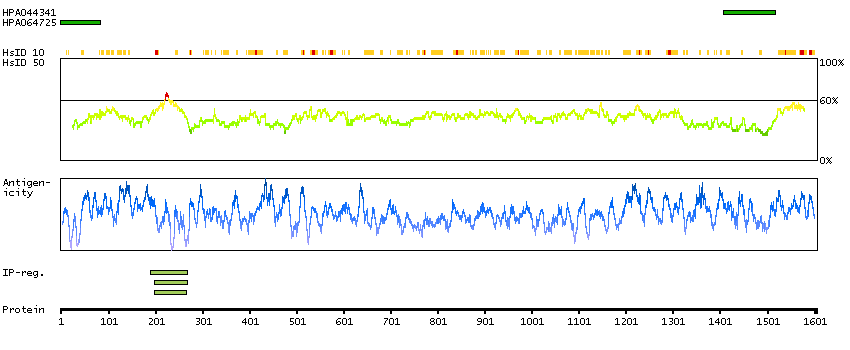
The protein encoded by this gene is an ortholog of the Drosophila melanogaster capicua gene, and is a member of the high mobility group (HMG)-box superfamily of transcriptional repressors. This protein contains a conserved HMG domain that is involved in DNA binding and nuclear localization, and a conserved C-terminus. Studies suggest that the N-terminal region of this protein interacts with Atxn1 (GeneID:6310), to form a transcription repressor complex, and in vitro studies suggest that polyglutamine-expansion of ATXN1 may alter the repressor activity of this complex. Mutations in this gene have been associated with olidogdendrogliomas (PMID:21817013). In addition, translocation events resulting in gene fusions of this gene with both DUX4 (GeneID:100288687) and FOXO4 (GeneID:4303) have been associated with round cell sarcomas. There are multiple pseudogenes of this gene found on chromosomes 1, 4, 6, 7, 16, 20, and the Y chromosome. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Feb 2015]
Predicted intracellular proteins Transcription factors Other all-alpha-helical DNA-binding domains Cancer-related genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Somatic Mutations COSMIC Missense Mutations COSMIC Frameshift Mutations COSMIC Translocations Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)