We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
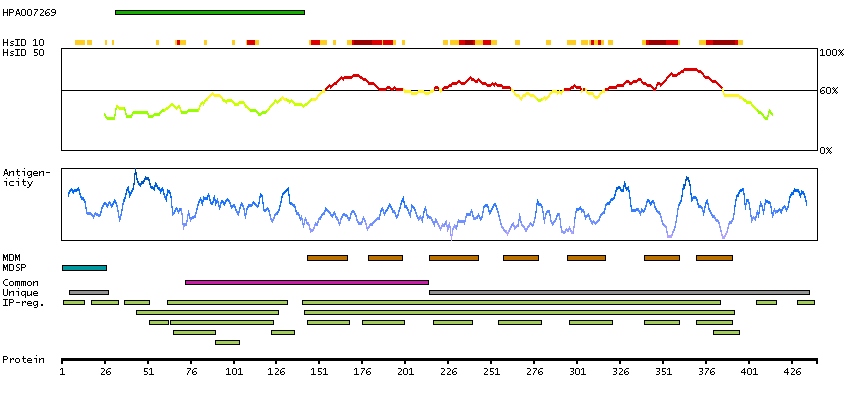
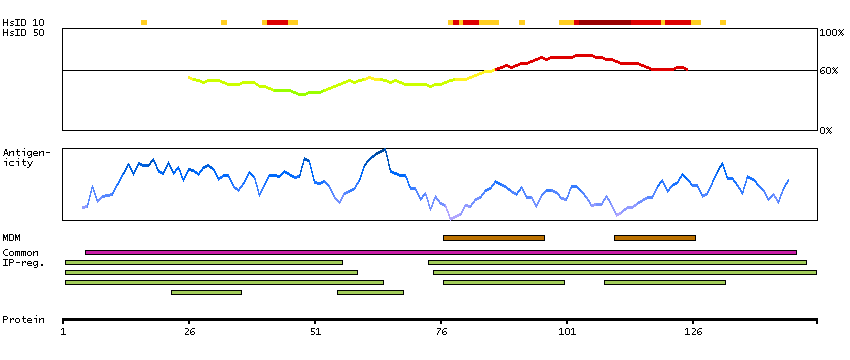
The protein encoded by this gene is a G protein-coupled receptor and belongs to the glucagon-VIP-secretin receptor family. It binds secretin which is the most potent regulator of pancreatic bicarbonate, electrolyte and volume secretion. Secretin and its receptor are suggested to be involved in pancreatic cancer and autism. [provided by RefSeq, Jul 2008]