We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
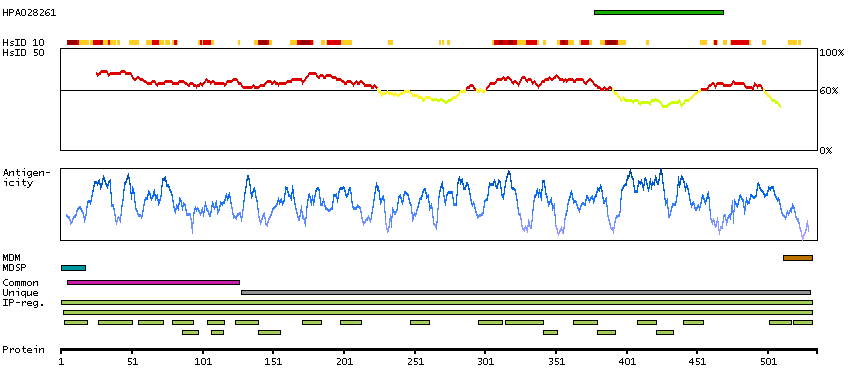
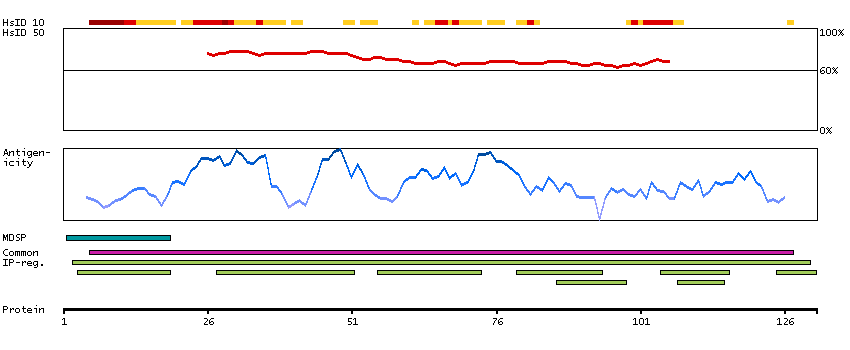
This gene encodes a flavin-containing monooxygenase family member. It is an NADPH-dependent enzyme that catalyzes the N-oxidation of some primary alkylamines through an N-hydroxylamine intermediate. However, some human populations contain an allele (FMO2*2A) with a premature stop codon, resulting in a protein that is C-terminally-truncated, has no catalytic activity, and is likely degraded rapidly. This gene is found in a cluster with other related family members on chromosome 1. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Aug 2014]