We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
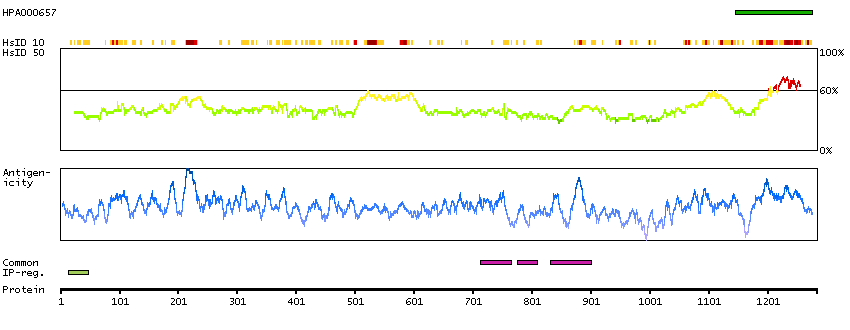
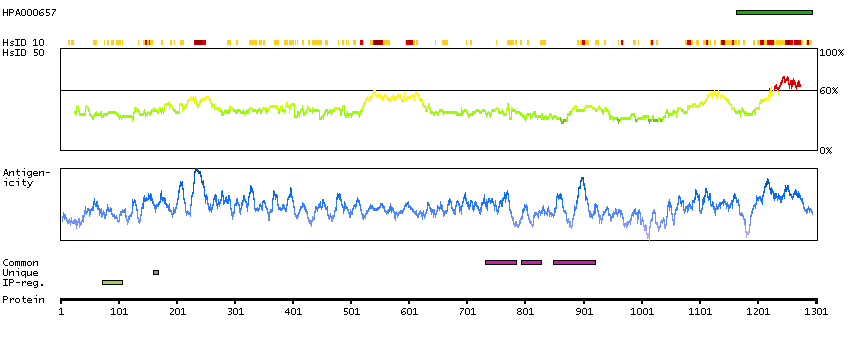
Apoptosis is defined by several morphologic nuclear changes, including chromatin condensation and nuclear fragmentation. This gene encodes a nuclear protein that induces apoptotic chromatin condensation after activation by caspase-3, without inducing DNA fragmentation. This protein has also been shown to be a component of a splicing-dependent multiprotein exon junction complex (EJC) that is deposited at splice junctions on mRNAs, as a consequence of pre-mRNA splicing. It may thus be involved in mRNA metabolism associated with splicing. Alternatively spliced transcript variants encoding different isoforms have been described for this gene. [provided by RefSeq, Oct 2011]
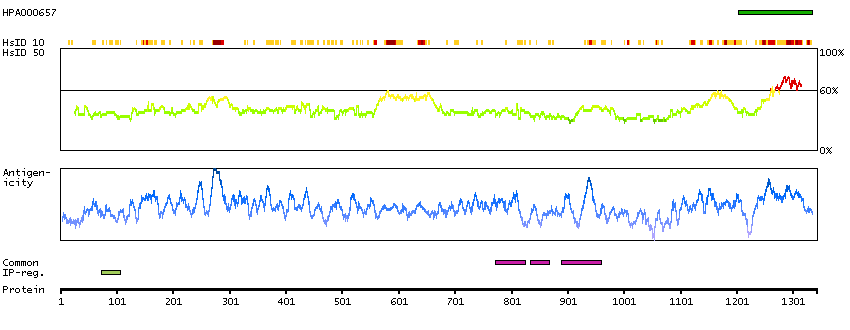
Q9UKV3 [Direct mapping] Apoptotic chromatin condensation inducer in the nucleus
Show all
Transporters Primary Active Transporters Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
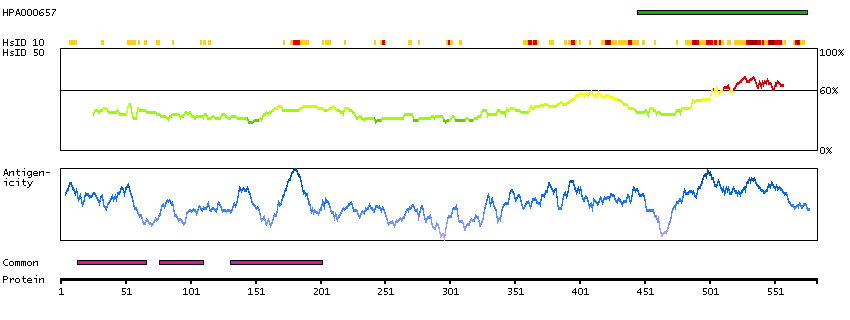
Q9UKV3 [Direct mapping] Apoptotic chromatin condensation inducer in the nucleus
Show all
Transporters Primary Active Transporters Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
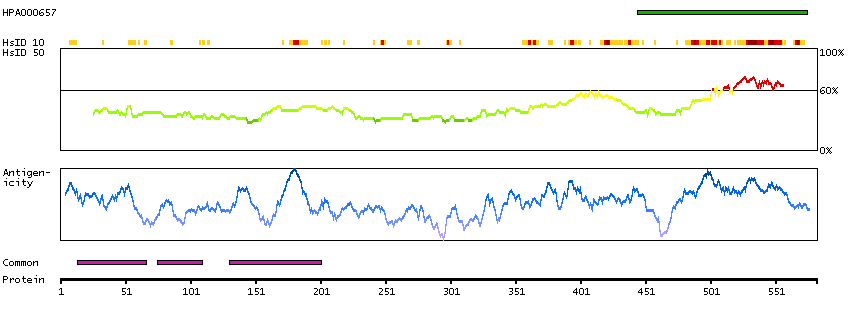
Q9UKV3 [Direct mapping] Apoptotic chromatin condensation inducer in the nucleus
Show all
Transporters Primary Active Transporters Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
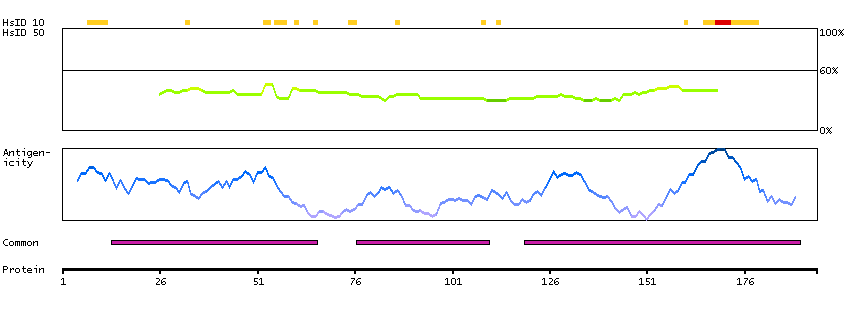
Q9UKV3 [Direct mapping] Apoptotic chromatin condensation inducer in the nucleus
Show all
Transporters Primary Active Transporters Predicted intracellular proteins Plasma proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)