We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
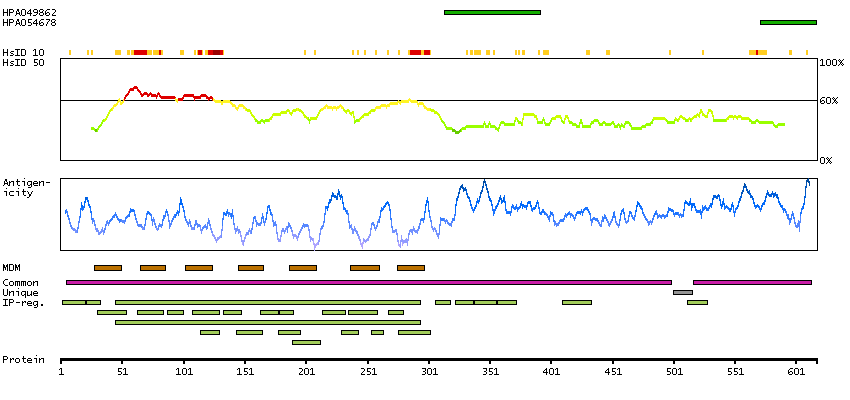
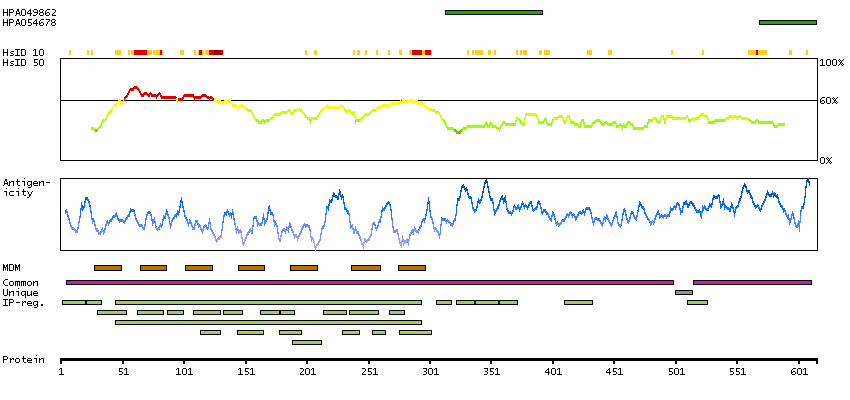
Presumed off target binding observed and disregarded. Antibody staining consistent with external characterization data and GTEx RNA expression data. Pending retesting.
This gene product belongs to the G-protein coupled receptor 1 family. Even though this protein shares similarity with the melatonin receptors, it does not bind melatonin, however, it inhibits melatonin receptor 1A function through heterodimerization. Polymorphic variants of this gene have been associated with bipolar affective disorder in women. [provided by RefSeq, Jan 2010]