We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
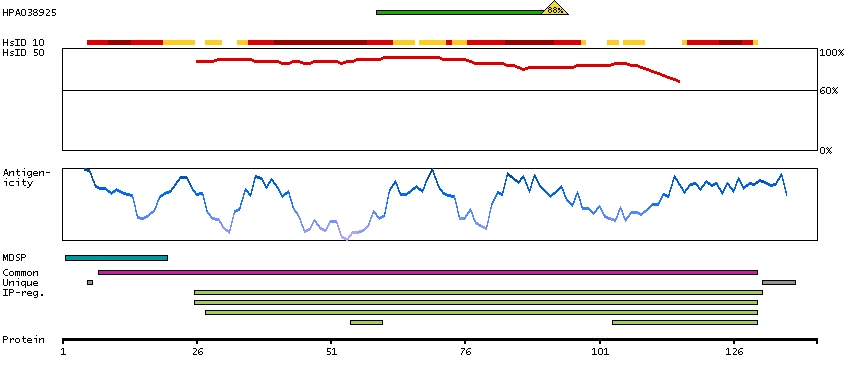
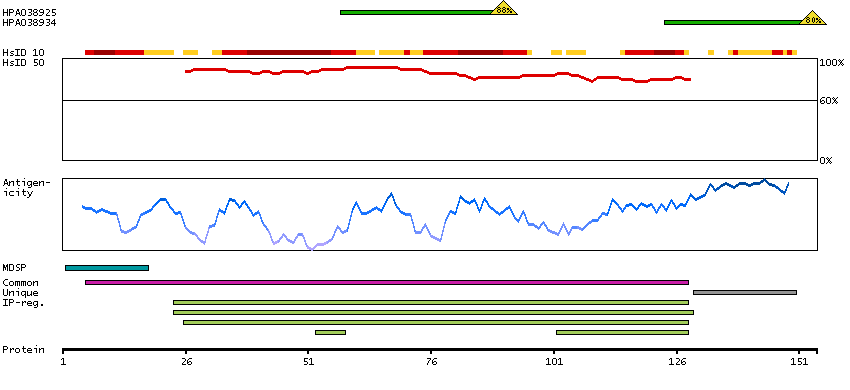
This gene is a member of the glycoprotein hormone beta chain family and encodes the beta subunit of luteinizing hormone (LH). Glycoprotein hormones are heterodimers consisting of a common alpha subunit and an unique beta subunit which confers biological specificity. LH is expressed in the pituitary gland and promotes spermatogenesis and ovulation by stimulating the testes and ovaries to synthesize steroids. The genes for the beta chains of chorionic gonadotropin and for luteinizing hormone are contiguous on chromosome 19q13.3. Mutations in this gene are associated with hypogonadism which is characterized by infertility and pseudohermaphroditism. [provided by RefSeq, Jul 2008]