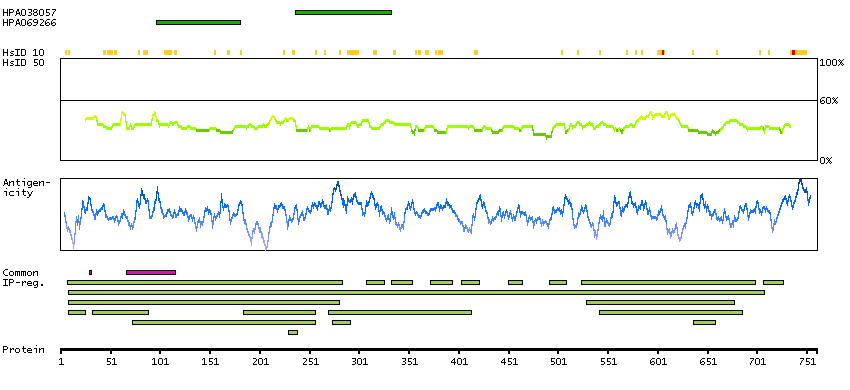
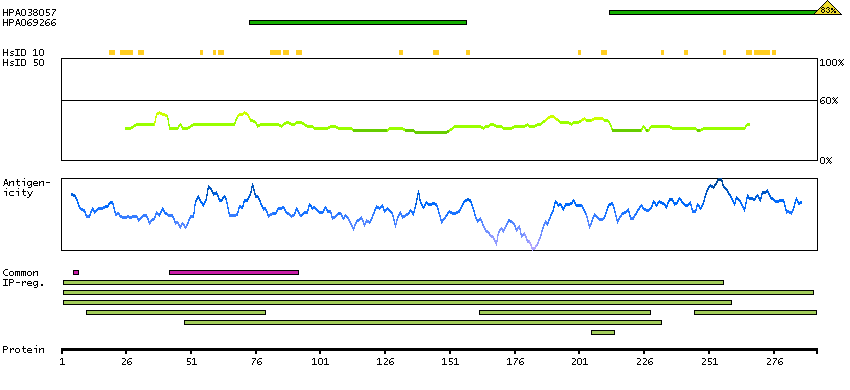
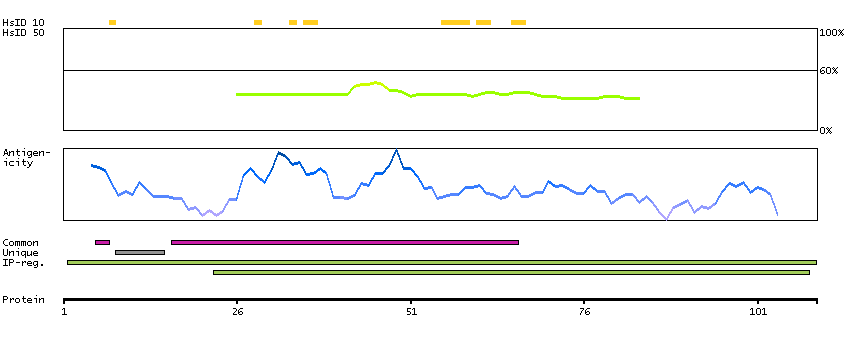
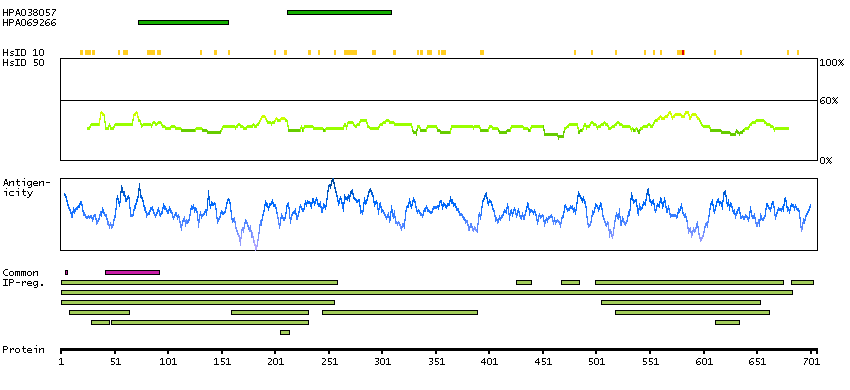
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
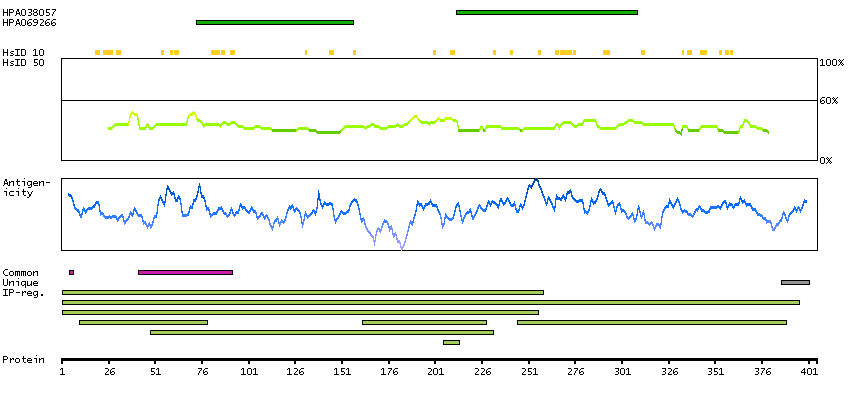
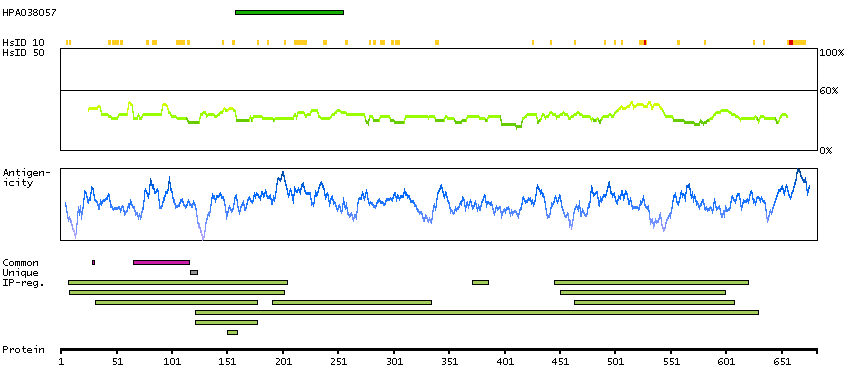
Excision repair cross-complementation group 2 (HGNC Symbol)
Entrez gene summary
The nucleotide excision repair pathway is a mechanism to repair damage to DNA. The protein encoded by this gene is involved in transcription-coupled nucleotide excision repair and is an integral member of the basal transcription factor BTF2/TFIIH complex. The gene product has ATP-dependent DNA helicase activity and belongs to the RAD3/XPD subfamily of helicases. Defects in this gene can result in three different disorders, the cancer-prone syndrome xeroderma pigmentosum complementation group D, trichothiodystrophy, and Cockayne syndrome. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Aug 2008]
Enzymes ENZYME proteins Hydrolases THUMBUP predicted membrane proteins Predicted intracellular proteins Cancer-related genes Candidate cancer biomarkers Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Splicing Mutations COSMIC Nonsense Mutations COSMIC Missense Mutations COSMIC Germline Mutations COSMIC Frameshift Mutations Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003676 [nucleic acid binding] GO:0003677 [DNA binding] GO:0004003 [ATP-dependent DNA helicase activity] GO:0004672 [protein kinase activity] GO:0005515 [protein binding] GO:0005524 [ATP binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005675 [holo TFIIH complex] GO:0005737 [cytoplasm] GO:0005819 [spindle] GO:0006281 [DNA repair] GO:0006283 [transcription-coupled nucleotide-excision repair] GO:0006289 [nucleotide-excision repair] GO:0006360 [transcription from RNA polymerase I promoter] GO:0006361 [transcription initiation from RNA polymerase I promoter] GO:0006362 [transcription elongation from RNA polymerase I promoter] GO:0006363 [termination of RNA polymerase I transcription] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006367 [transcription initiation from RNA polymerase II promoter] GO:0006368 [transcription elongation from RNA polymerase II promoter] GO:0006370 [7-methylguanosine mRNA capping] GO:0006468 [protein phosphorylation] GO:0006915 [apoptotic process] GO:0006979 [response to oxidative stress] GO:0007059 [chromosome segregation] GO:0008022 [protein C-terminus binding] GO:0008026 [ATP-dependent helicase activity] GO:0008094 [DNA-dependent ATPase activity] GO:0008353 [RNA polymerase II carboxy-terminal domain kinase activity] GO:0009650 [UV protection] GO:0010467 [gene expression] GO:0016032 [viral process] GO:0016818 [hydrolase activity, acting on acid anhydrides, in phosphorus-containing anhydrides] GO:0019907 [cyclin-dependent protein kinase activating kinase holoenzyme complex] GO:0032508 [DNA duplex unwinding] GO:0033683 [nucleotide-excision repair, DNA incision] GO:0035315 [hair cell differentiation] GO:0040029 [regulation of gene expression, epigenetic] GO:0043139 [5'-3' DNA helicase activity] GO:0044281 [small molecule metabolic process] GO:0045814 [negative regulation of gene expression, epigenetic] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046872 [metal ion binding] GO:0047485 [protein N-terminus binding] GO:0050434 [positive regulation of viral transcription] GO:0051539 [4 iron, 4 sulfur cluster binding] GO:0070911 [global genome nucleotide-excision repair] GO:0071817 [MMXD complex] GO:1901990 [regulation of mitotic cell cycle phase transition]