We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
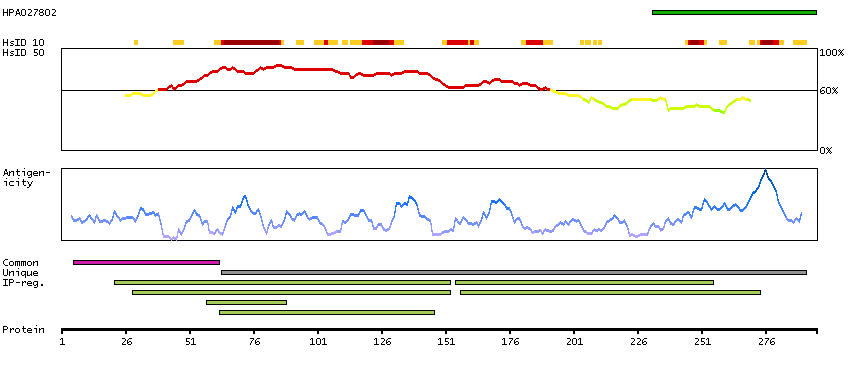
The protein encoded by this gene belongs to the highly conserved cyclin family, whose members are characterized by a dramatic periodicity in protein abundance throughout the cell cycle. Cyclins function as regulators of CDK kinases. Different cyclins exhibit distinct expression and degradation patterns which contribute to the temporal coordination of each mitotic event. This cyclin forms a complex with and functions as a regulatory subunit of CDK4 or CDK6, whose activity is required for cell cycle G1/S transition. This protein has been shown to interact with tumor suppressor protein Rb and the expression of this gene is regulated positively by Rb. Mutations, amplification and overexpression of this gene, which alters cell cycle progression, are observed frequently in a variety of tumors and may contribute to tumorigenesis. [provided by RefSeq, Jul 2008]
SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Cancer-related genes Candidate cancer biomarkers Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes FDA approved drug targets Small molecule drugs Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000082 [G1/S transition of mitotic cell cycle] GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000278 [mitotic cell cycle] GO:0000307 [cyclin-dependent protein kinase holoenzyme complex] GO:0000320 [re-entry into mitotic cell cycle] GO:0001889 [liver development] GO:0001934 [positive regulation of protein phosphorylation] GO:0003714 [transcription corepressor activity] GO:0004672 [protein kinase activity] GO:0005515 [protein binding] GO:0005622 [intracellular] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0005923 [bicellular tight junction] GO:0006325 [chromatin organization] GO:0006351 [transcription, DNA-templated] GO:0006468 [protein phosphorylation] GO:0006974 [cellular response to DNA damage stimulus] GO:0007219 [Notch signaling pathway] GO:0007595 [lactation] GO:0008134 [transcription factor binding] GO:0008284 [positive regulation of cell proliferation] GO:0010033 [response to organic substance] GO:0010039 [response to iron ion] GO:0010165 [response to X-ray] GO:0010243 [response to organonitrogen compound] GO:0010971 [positive regulation of G2/M transition of mitotic cell cycle] GO:0014070 [response to organic cyclic compound] GO:0016020 [membrane] GO:0016055 [Wnt signaling pathway] GO:0016301 [kinase activity] GO:0016538 [cyclin-dependent protein serine/threonine kinase regulator activity] GO:0017053 [transcriptional repressor complex] GO:0019899 [enzyme binding] GO:0019901 [protein kinase binding] GO:0030178 [negative regulation of Wnt signaling pathway] GO:0030857 [negative regulation of epithelial cell differentiation] GO:0030968 [endoplasmic reticulum unfolded protein response] GO:0031100 [organ regeneration] GO:0031571 [mitotic G1 DNA damage checkpoint] GO:0032026 [response to magnesium ion] GO:0032355 [response to estradiol] GO:0032403 [protein complex binding] GO:0033197 [response to vitamin E] GO:0033327 [Leydig cell differentiation] GO:0033598 [mammary gland epithelial cell proliferation] GO:0033601 [positive regulation of mammary gland epithelial cell proliferation] GO:0042493 [response to drug] GO:0042826 [histone deacetylase binding] GO:0043627 [response to estrogen] GO:0045444 [fat cell differentiation] GO:0045471 [response to ethanol] GO:0045737 [positive regulation of cyclin-dependent protein serine/threonine kinase activity] GO:0048545 [response to steroid hormone] GO:0051301 [cell division] GO:0051384 [response to glucocorticoid] GO:0051412 [response to corticosterone] GO:0051592 [response to calcium ion] GO:0051726 [regulation of cell cycle] GO:0060070 [canonical Wnt signaling pathway] GO:0060749 [mammary gland alveolus development] GO:0070064 [proline-rich region binding] GO:0070141 [response to UV-A] GO:0071157 [negative regulation of cell cycle arrest] GO:0071310 [cellular response to organic substance] GO:2000045 [regulation of G1/S transition of mitotic cell cycle]
Predicted intracellular proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)