We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Cytoplasmic positivity in renal tubules and hepatocytes.
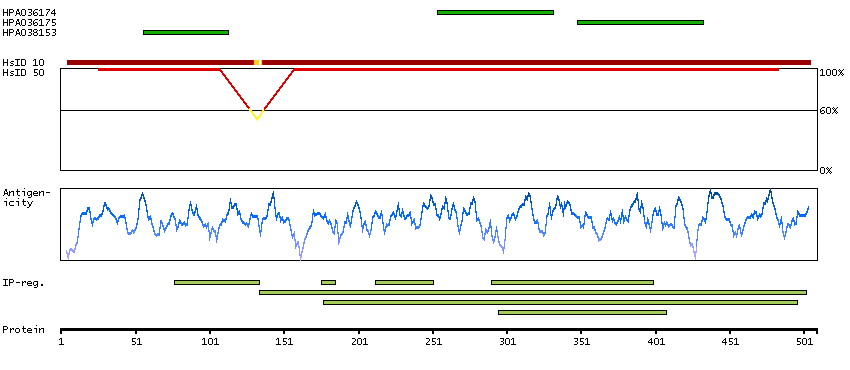
DATA RELIABILITY
Data reliability description
No internal RNA expression data available for correlation. Caution, targets protein from more than one gene. Presumed off target binding observed and disregarded.
This locus represents naturally occurring read-through transcription between the neighboring abhydrolase domain containing 14A (ABHD14A) and aminoacylase 1 (ACY1) genes on chromosome 3. The read-through transcript encodes a protein that shares sequence identity with the downstream gene product but its N-terminal region is distinct due to the use of an alternate start codon relative to the upstream gene. [provided by RefSeq, Oct 2015]