We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
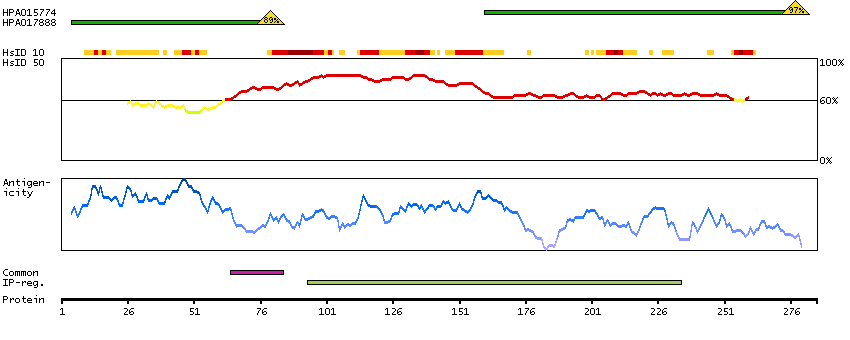
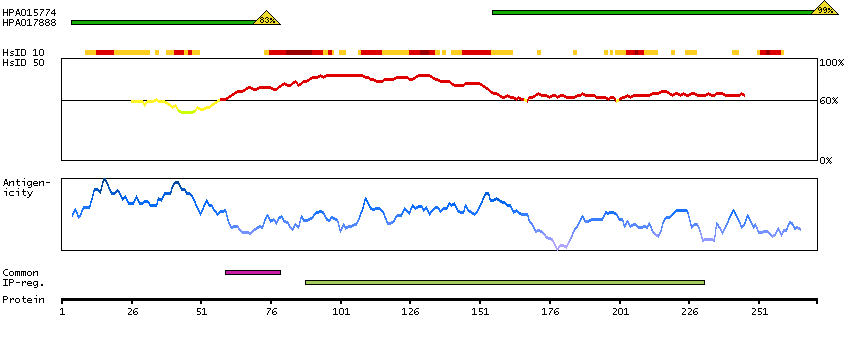
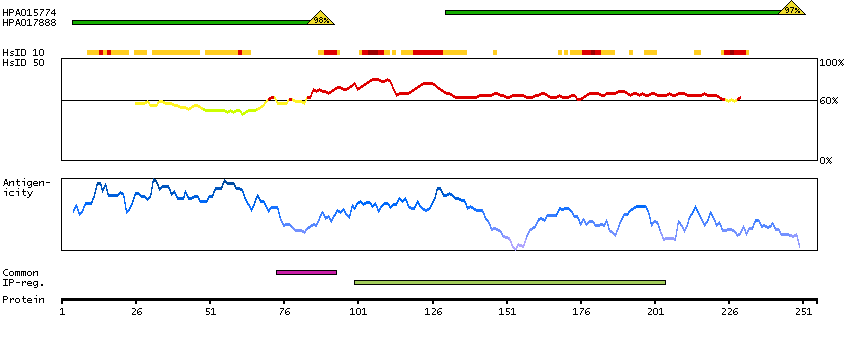
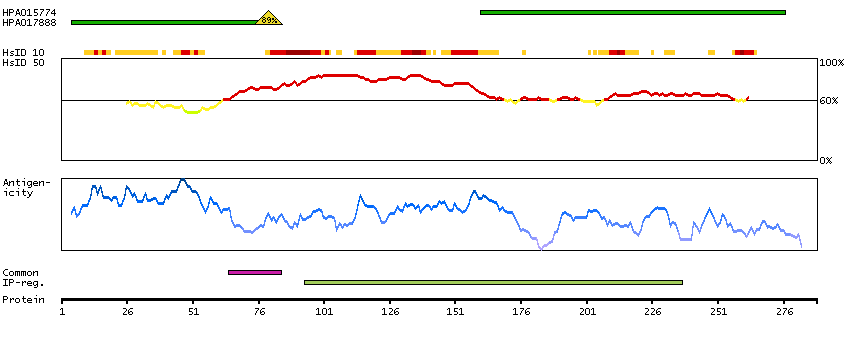
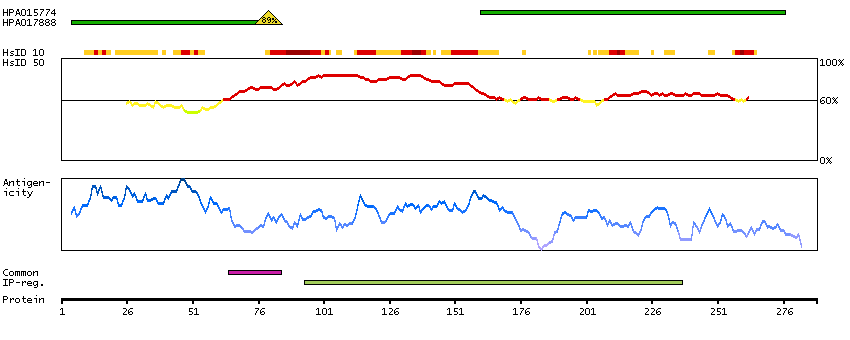
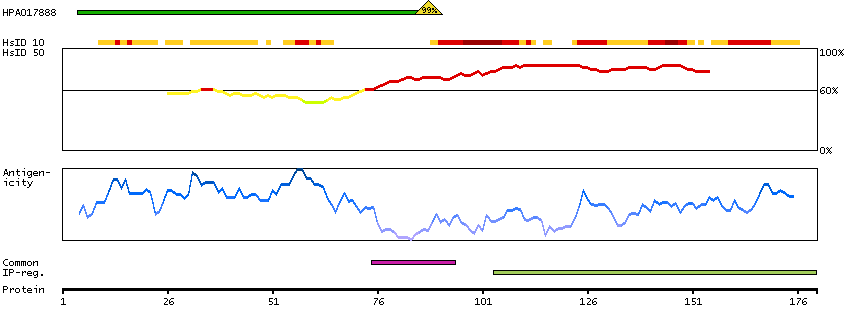
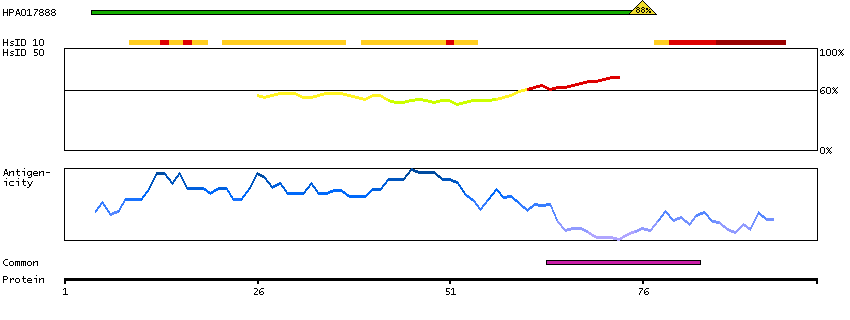
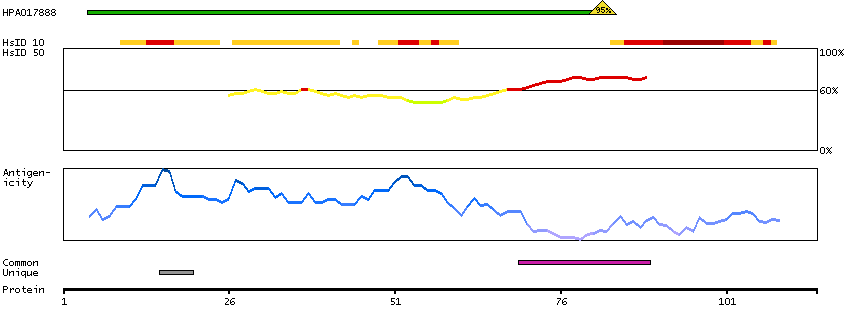
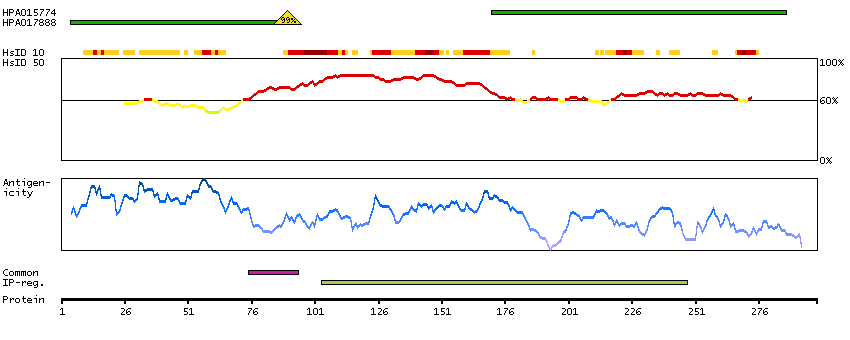
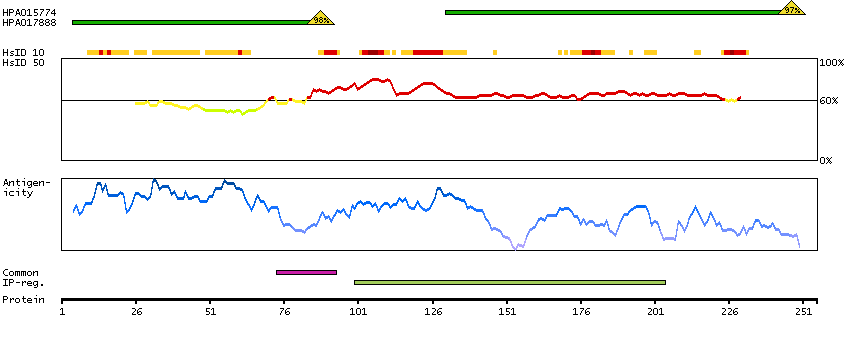
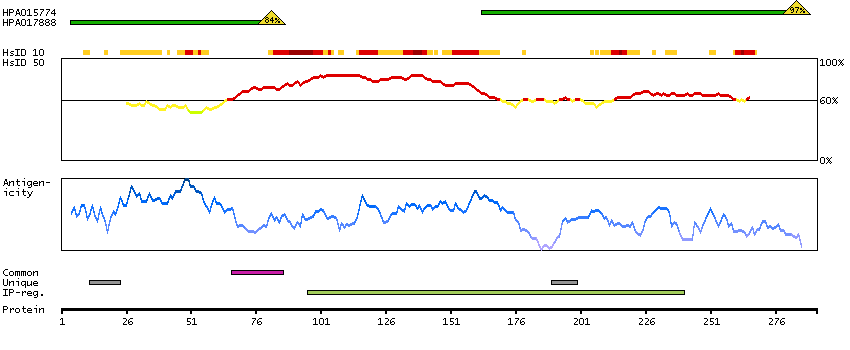
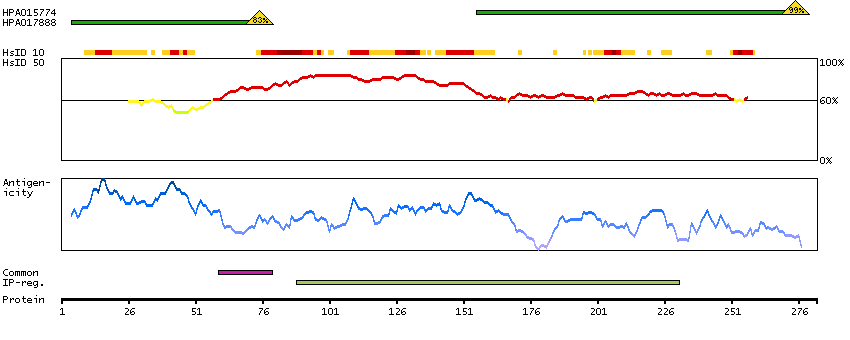
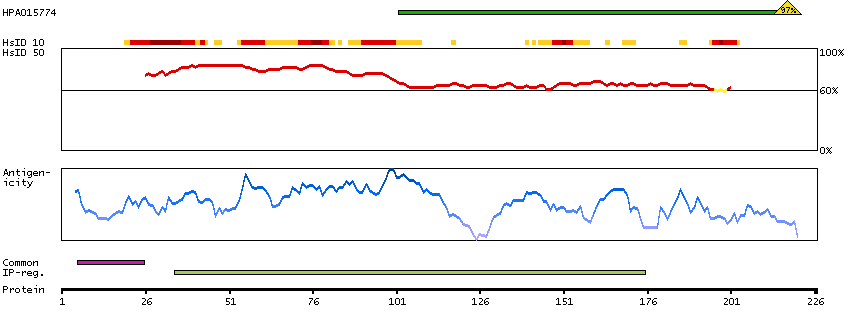
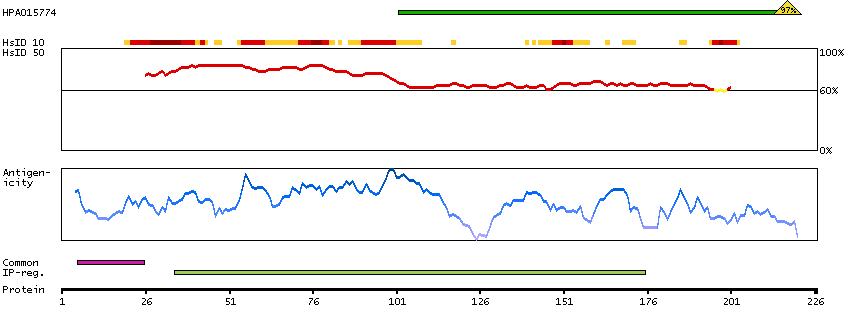
The protein encoded by this gene is the tropomyosin-binding subunit of the troponin complex, which is located on the thin filament of striated muscles and regulates muscle contraction in response to alterations in intracellular calcium ion concentration. Mutations in this gene have been associated with familial hypertrophic cardiomyopathy as well as with dilated cardiomyopathy. Transcripts for this gene undergo alternative splicing that results in many tissue-specific isoforms, however, the full-length nature of some of these variants has not yet been determined. [provided by RefSeq, Jul 2008]
Predicted intracellular proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Predicted intracellular proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Predicted intracellular proteins Candidate cardiovascular disease genes Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)