We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
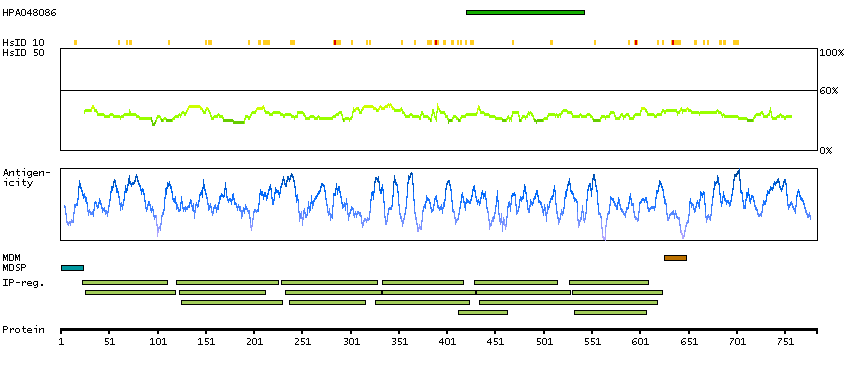
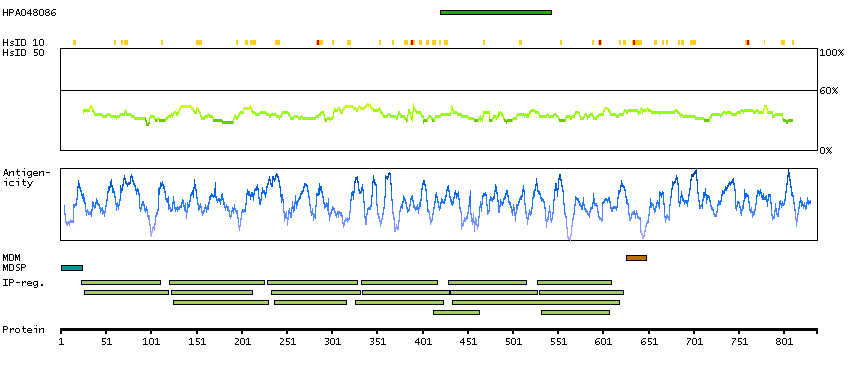
The protein encoded by this gene is the receptor for colony stimulating factor 3, a cytokine that controls the production, differentiation, and function of granulocytes. The encoded protein, which is a member of the family of cytokine receptors, may also function in some cell surface adhesion or recognition processes. Alternatively spliced transcript variants have been described. Mutations in this gene are a cause of Kostmann syndrome, also known as severe congenital neutropenia. [provided by RefSeq, Aug 2010]