We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
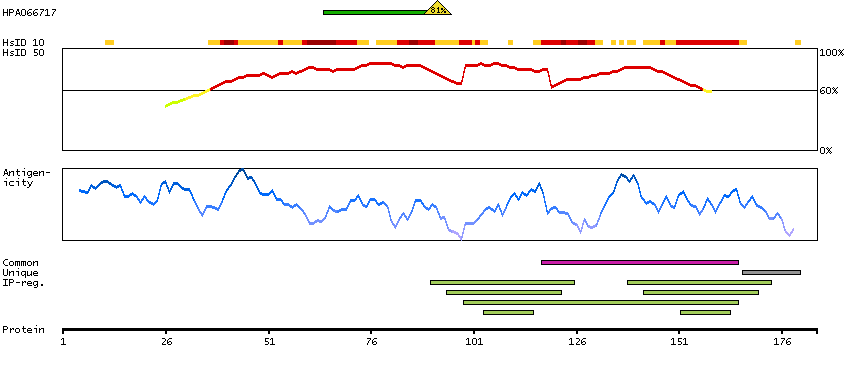
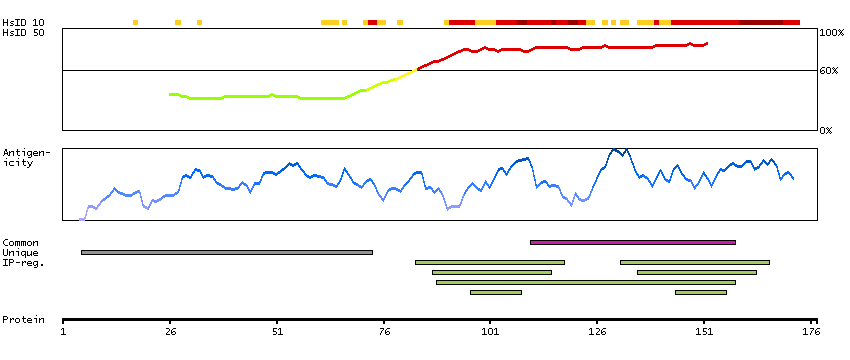
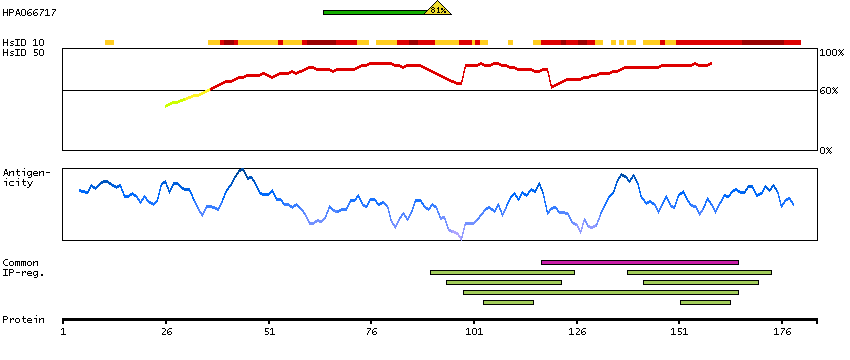
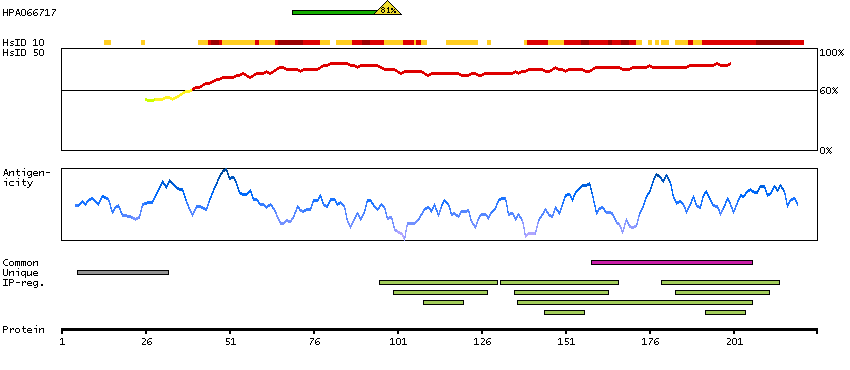
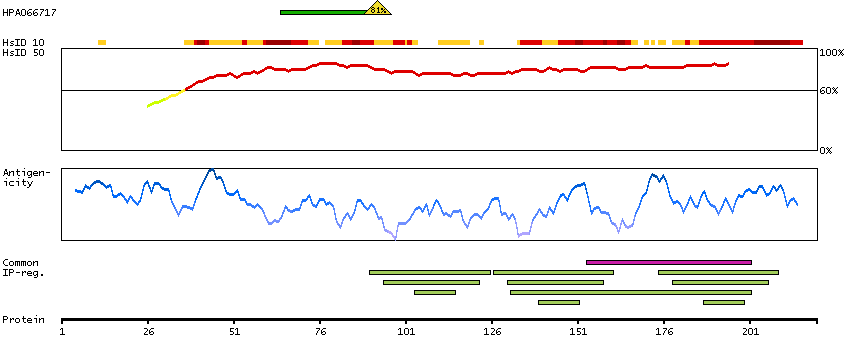
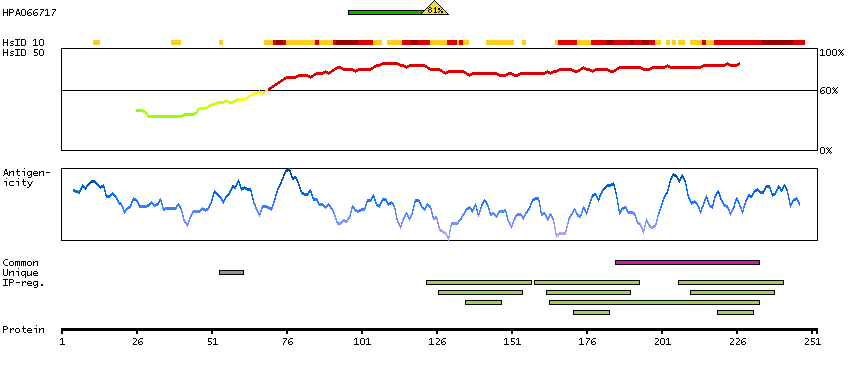
This gene encodes a member of the family of voltage-gated potassium (Kv) channel-interacting proteins (KCNIPs), which belongs to the recoverin branch of the EF-hand superfamily. Members of the KCNIP family are small calcium binding proteins. They all have EF-hand-like domains, and differ from each other in the N-terminus. They are integral subunit components of native Kv4 channel complexes. They may regulate A-type currents, and hence neuronal excitability, in response to changes in intracellular calcium. Multiple alternatively spliced transcript variants encoding distinct isoforms have been identified from this gene. [provided by RefSeq, Jul 2008]