We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
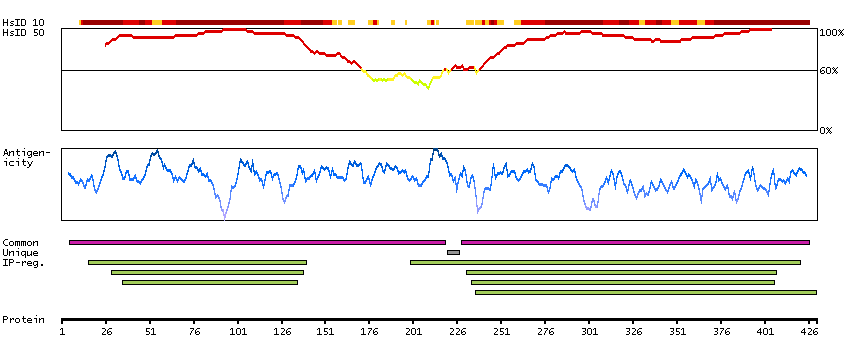
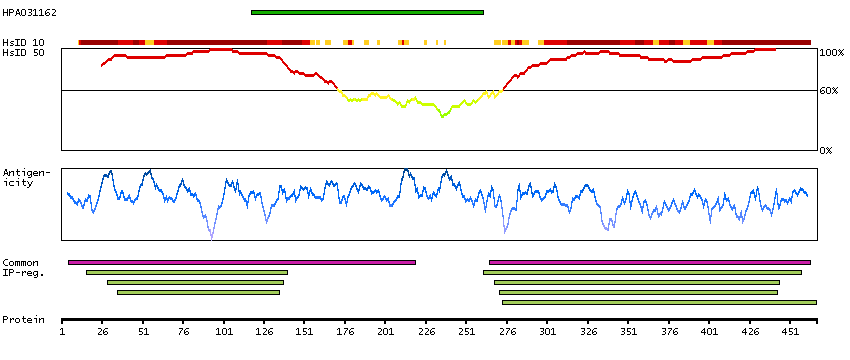
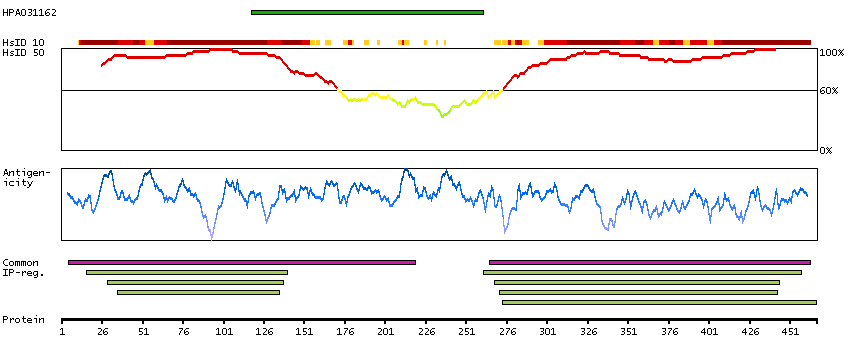
The protein encoded by this gene is a member of the SMAD family, which transduces signals from TGF-beta family members. The encoded protein is activated by bone morphogenetic proteins and interacts with SMAD4. Two transcript variants encoding different isoforms have been found for this gene.[provided by RefSeq, Jan 2010]
O15198 [Direct mapping] Mothers against decapentaplegic homolog 9 A0A024RDR3 [Target identity:100%; Query identity:100%] Mothers against decapentaplegic homolog
Show all
Predicted intracellular proteins Transcription factors beta-Hairpin exposed by an alpha/beta-scaffold Disease related genes Protein evidence (Kim et al 2014)
Show all
GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005622 [intracellular] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0007179 [transforming growth factor beta receptor signaling pathway] GO:0030509 [BMP signaling pathway] GO:0030618 [transforming growth factor beta receptor, pathway-specific cytoplasmic mediator activity] GO:0035556 [intracellular signal transduction] GO:0046872 [metal ion binding] GO:0060395 [SMAD protein signal transduction] GO:0071141 [SMAD protein complex] GO:0071773 [cellular response to BMP stimulus] GO:1901522 [positive regulation of transcription from RNA polymerase II promoter involved in cellular response to chemical stimulus]
O15198 [Direct mapping] Mothers against decapentaplegic homolog 9
Show all
Predicted intracellular proteins Transcription factors beta-Hairpin exposed by an alpha/beta-scaffold Disease related genes Protein evidence (Kim et al 2014)
Show all
GO:0001657 [ureteric bud development] GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005622 [intracellular] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0006351 [transcription, DNA-templated] GO:0006355 [regulation of transcription, DNA-templated] GO:0006468 [protein phosphorylation] GO:0007179 [transforming growth factor beta receptor signaling pathway] GO:0030509 [BMP signaling pathway] GO:0030618 [transforming growth factor beta receptor, pathway-specific cytoplasmic mediator activity] GO:0030901 [midbrain development] GO:0030902 [hindbrain development] GO:0035556 [intracellular signal transduction] GO:0046872 [metal ion binding] GO:0051216 [cartilage development] GO:0060348 [bone development] GO:0060395 [SMAD protein signal transduction] GO:0071141 [SMAD protein complex] GO:0071407 [cellular response to organic cyclic compound] GO:0071773 [cellular response to BMP stimulus] GO:1901522 [positive regulation of transcription from RNA polymerase II promoter involved in cellular response to chemical stimulus]