We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
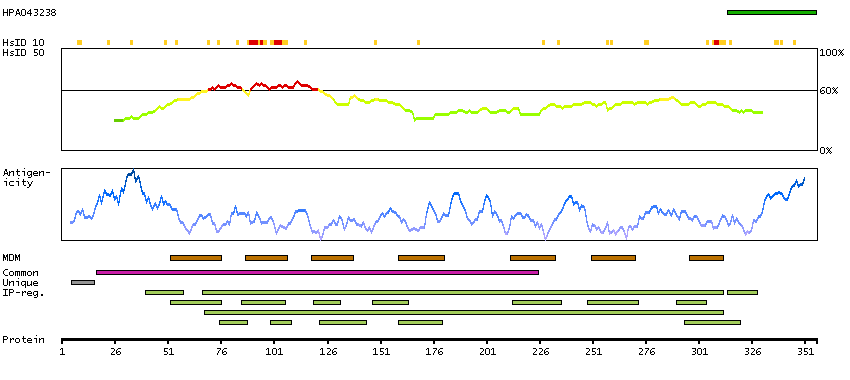
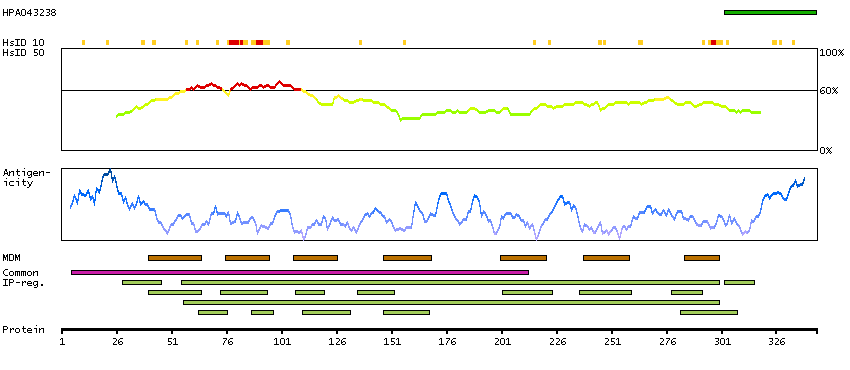
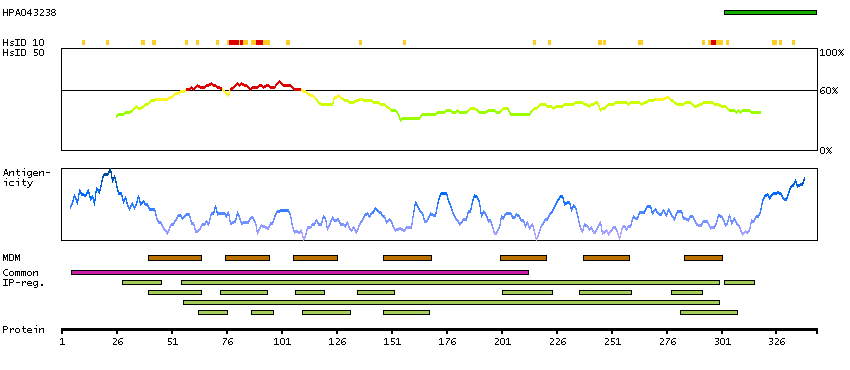
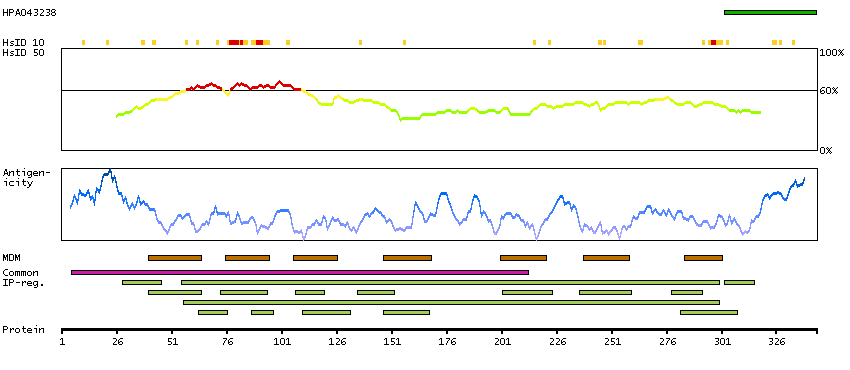
This gene encodes a chemokine receptor like protein, which is predicted to be a seven transmembrane protein and most closely related to CCR1. Chemokines and their receptors mediated signal transduction are critical for the recruitment of effector immune cells to the site of inflammation. This gene is expressed at high levels in primary neutrophils and primary monocytes, and is further upregulated on neutrophil activation and during monocyte to macrophage differentiation. The function of this gene is unknown. This gene is mapped to the region where the chemokine receptor gene cluster is located. [provided by RefSeq, Jul 2008]