We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
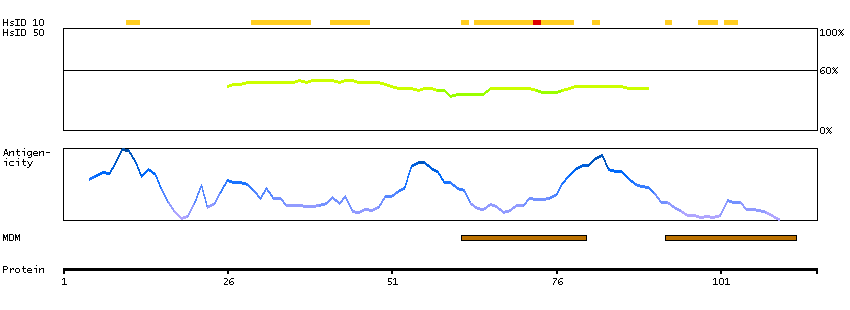
This gene encodes a member of the family of adenylate cyclases, which are membrane-associated enzymes that catalyze the formation of the secondary messenger cyclic adenosine monophosphate (cAMP). Mouse studies show that adenylate cyclase 4, along with adenylate cyclases 2 and 3, is expressed in olfactory cilia, suggesting that several different adenylate cyclases may couple to olfactory receptors and that there may be multiple receptor-mediated mechanisms for the generation of cAMP signals. Alternative splicing results in transcript variants. [provided by RefSeq, Nov 2010]