We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
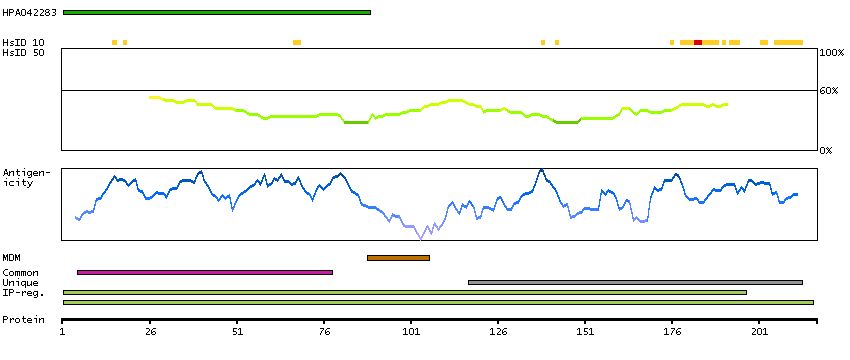
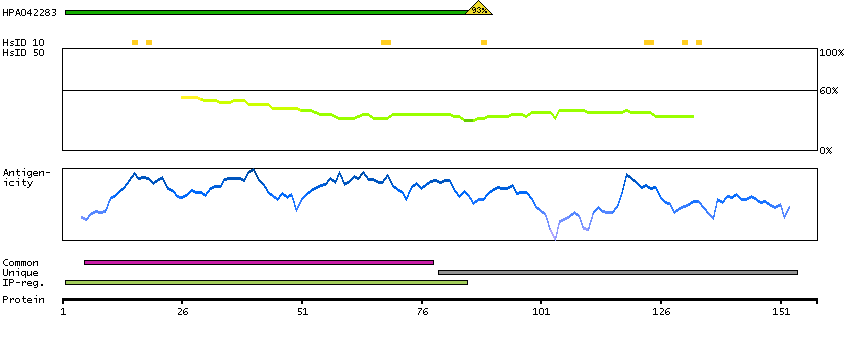
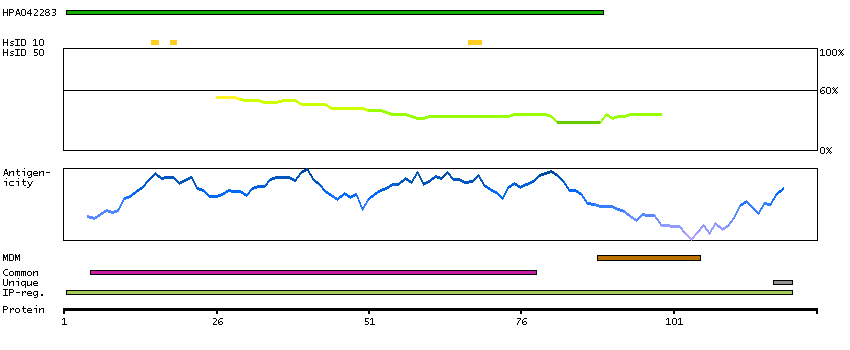
Calcyon neuron-specific vesicular protein (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a type II single transmembrane protein. It is required for maximal stimulated calcium release after stimulation of purinergic or muscarinic but not beta-adrenergic receptors. The encoded protein interacts with D1 dopamine receptor and may interact with other DA receptor subtypes and/or GPCRs. [provided by RefSeq, Jul 2008]