We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Solute carrier family 12 (potassium/chloride transporter), member 6 (HGNC Symbol)
Entrez gene summary
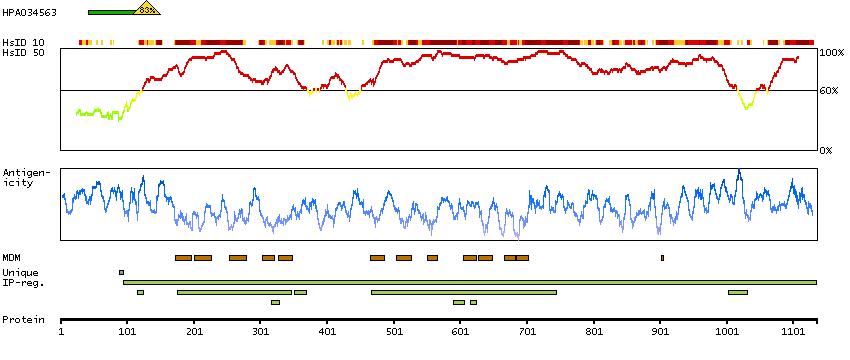
This gene is a member of the K-Cl cotransporter (KCC) family. K-Cl cotransporters are integral membrane proteins that lower intracellular chloride concentrations below the electrochemical equilibrium potential. The proteins encoded by this gene are activated by cell swelling induced by hypotonic conditions. Alternate splicing results in multiple transcript variants encoding different isoforms. Mutations in this gene are associated with agenesis of the corpus callosum with peripheral neuropathy. [provided by RefSeq, Jul 2008]