We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Disease related genes FDA approved drug targets Plasma proteins Predicted intracellular proteins Predicted membrane proteins Transporters Voltage-gated ion channels
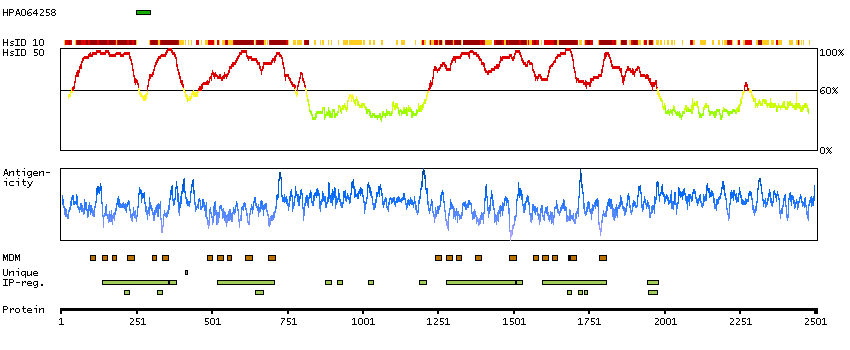
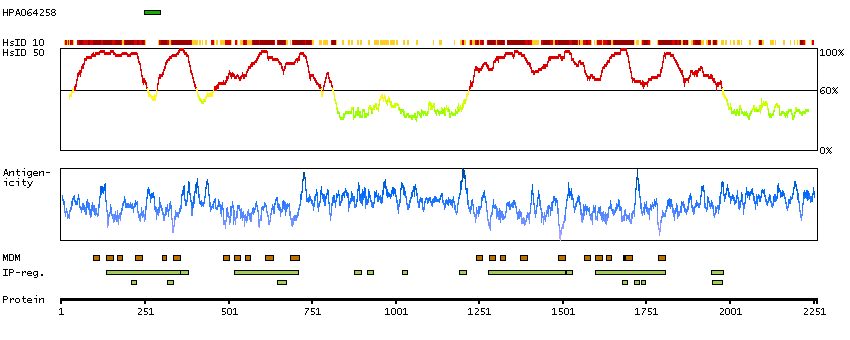
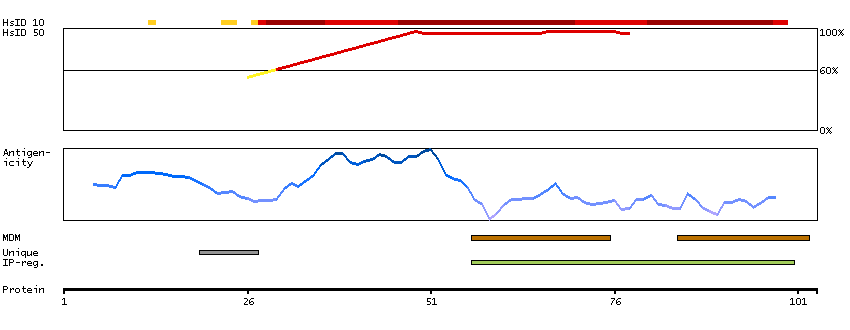
Voltage-dependent calcium channels mediate the entry of calcium ions into excitable cells, and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, and gene expression. Calcium channels are multisubunit complexes composed of alpha-1, beta, alpha-2/delta, and gamma subunits. The channel activity is directed by the pore-forming alpha-1 subunit, whereas, the others act as auxiliary subunits regulating this activity. The distinctive properties of the calcium channel types are related primarily to the expression of a variety of alpha-1 isoforms, alpha-1A, B, C, D, E, and S. This gene encodes the alpha-1A subunit, which is predominantly expressed in neuronal tissue. Mutations in this gene are associated with 2 neurologic disorders, familial hemiplegic migraine and episodic ataxia 2. This gene also exhibits polymorphic variation due to (CAG)n-repeats. Multiple transcript variants encoding different isoforms have been found for this gene. In one set of transcript variants, the (CAG)n-repeats occur in the 3' UTR, and are not associated with any disease. But in another set of variants, an insertion extends the coding region to include the (CAG)n-repeats which encode a polyglutamine tract. Expansion of the (CAG)n-repeats from the normal 4-16 to 21-28 in the coding region is associated with spinocerebellar ataxia 6. [provided by RefSeq, Mar 2010]