We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
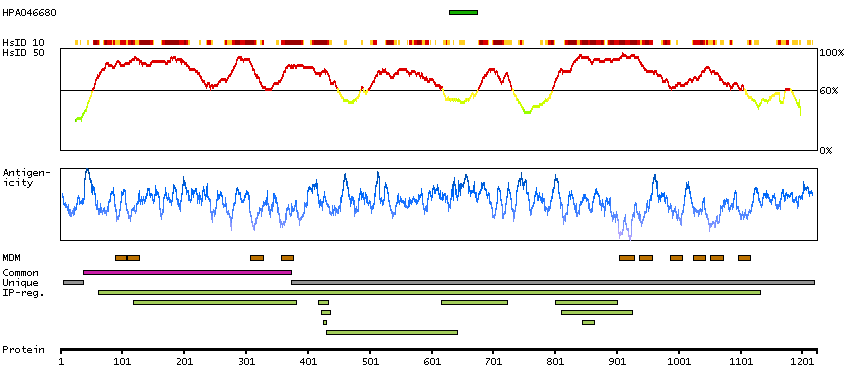
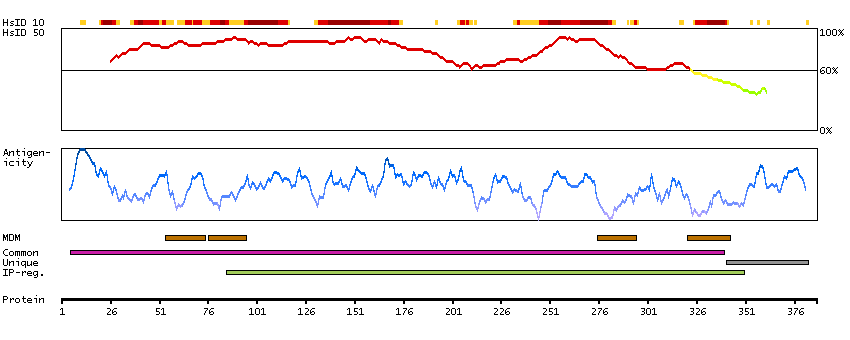
ATPase, aminophospholipid transporter, class I, type 8B, member 2 (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene belongs to the family of P-type cation transport ATPases, and to the subfamily of aminophospholipid-transporting ATPases. The aminophospholipid translocases transport phosphatidylserine and phosphatidylethanolamine from one side of a bilayer to another. Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]