We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (ovary, testis)
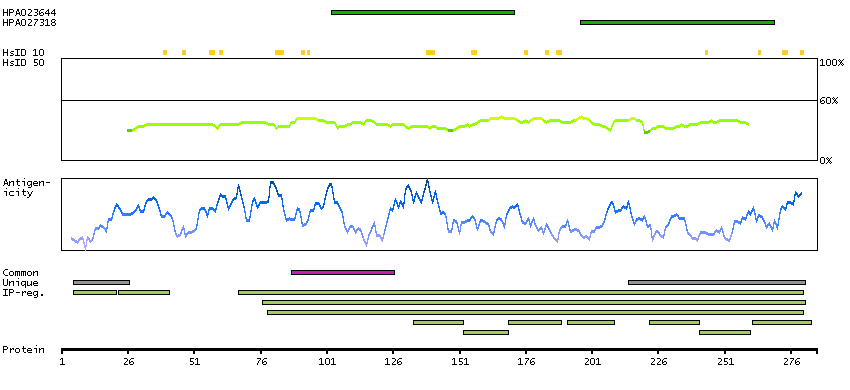
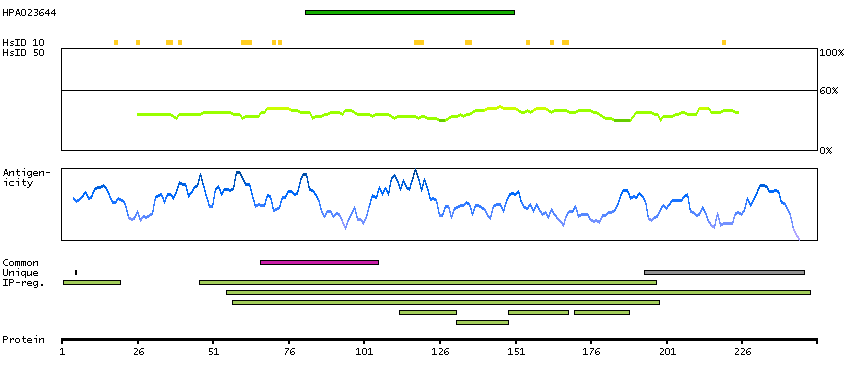
GENE INFORMATION
Gene name
STAR (HGNC Symbol)
Synonyms
StAR, STARD1
Description
Steroidogenic acute regulatory protein (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene plays a key role in the acute regulation of steroid hormone synthesis by enhancing the conversion of cholesterol into pregnenolone. This protein permits the cleavage of cholesterol into pregnenolone by mediating the transport of cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane. Mutations in this gene are a cause of congenital lipoid adrenal hyperplasia (CLAH), also called lipoid CAH. A pseudogene of this gene is located on chromosome 13. [provided by RefSeq, Jul 2008]