We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
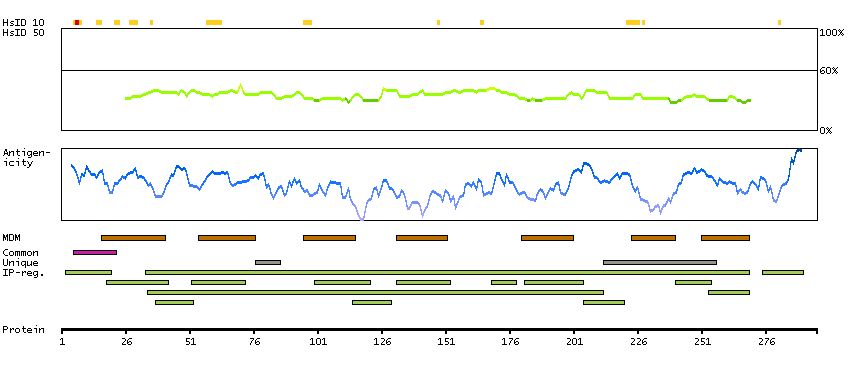
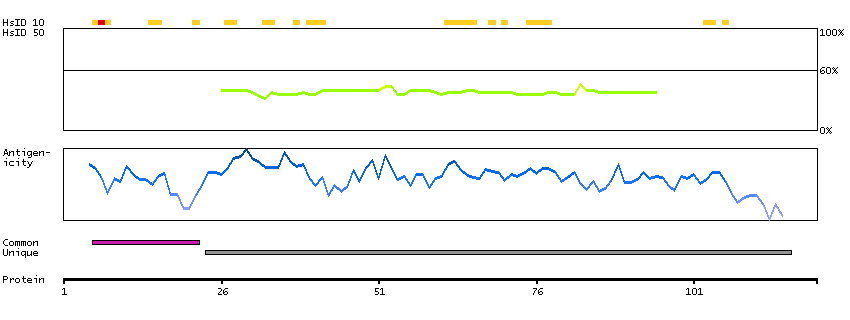
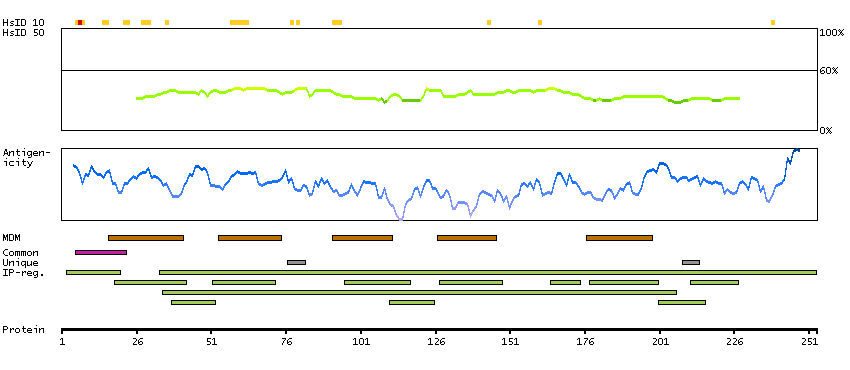
This gene encodes a putative retinal G-protein coupled receptor. The gene is a member of the opsin subfamily of the 7 transmembrane, G-protein coupled receptor 1 family. Like other opsins which bind retinaldehyde, it contains a conserved lysine residue in the seventh transmembrane domain. The protein acts as a photoisomerase to catalyze the conversion of all-trans-retinal to 11-cis-retinal. The reverse isomerization occurs with rhodopsin in retinal photoreceptor cells. The protein is exclusively expressed in tissue adjacent to retinal photoreceptor cells, the retinal pigment epithelium and Mueller cells. This gene may be associated with autosomal recessive and autosomal dominant retinitis pigmentosa (arRP and adRP, respectively). Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Jul 2008]