We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
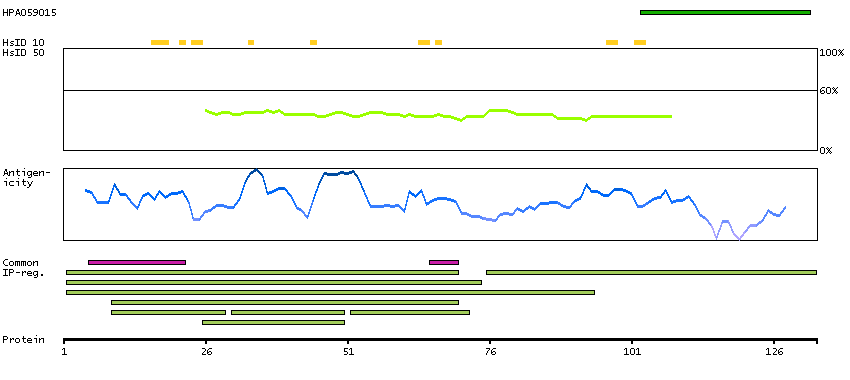
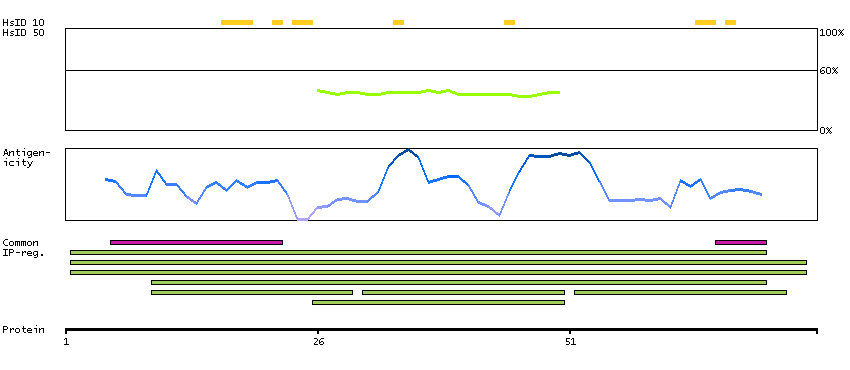
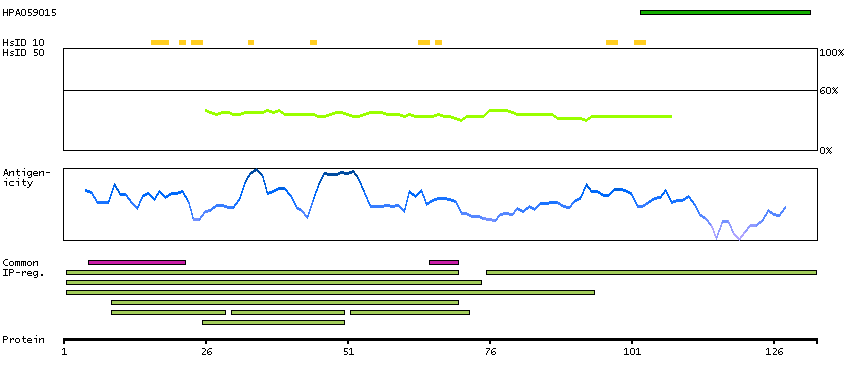
This gene is the cellular homolog of the fox sequence in the Finkel-Biskis-Reilly murine sarcoma virus (FBR-MuSV). It encodes a fusion protein consisting of the ubiquitin-like protein fubi at the N terminus and ribosomal protein S30 at the C terminus. It has been proposed that the fusion protein is post-translationally processed to generate free fubi and free ribosomal protein S30. Fubi is a member of the ubiquitin family, and ribosomal protein S30 belongs to the S30E family of ribosomal proteins. Whereas the function of fubi is currently unknown, ribosomal protein S30 is a component of the 40S subunit of the cytoplasmic ribosome and displays antimicrobial activity. Pseudogenes derived from this gene are present in the genome. Similar to ribosomal protein S30, ribosomal proteins S27a and L40 are synthesized as fusion proteins with ubiquitin. [provided by RefSeq, Nov 2014]