We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
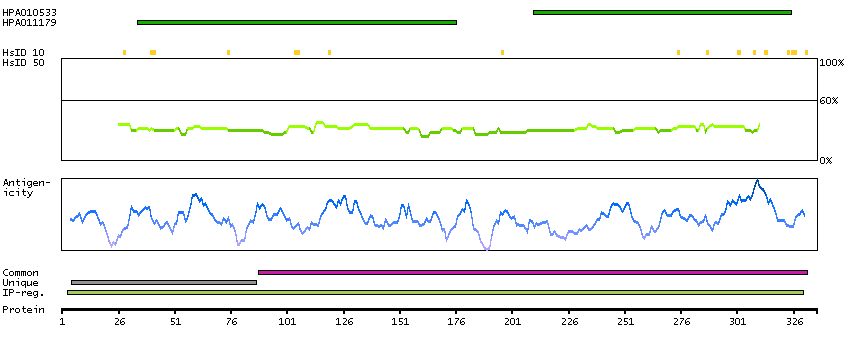
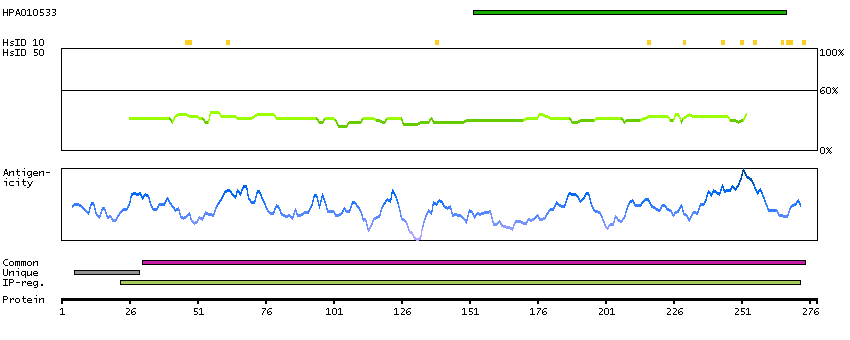
The neuronal excitotoxin quinolinate is an intermediate in the de novo synthesis pathway of NAD from tryptophan, and has been implicated in the pathogenesis of several neurodegenerative disorders. Quinolinate is derived from alpha-amino-beta-carboxy-muconate-epsilon-semialdehyde (ACMS). ACMSD (ACMS decarboxylase; EC 4.1.1.45) can divert ACMS to a benign catabolite and thus prevent the accumulation of quinolinate from ACMS.[supplied by OMIM, Oct 2004]