We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
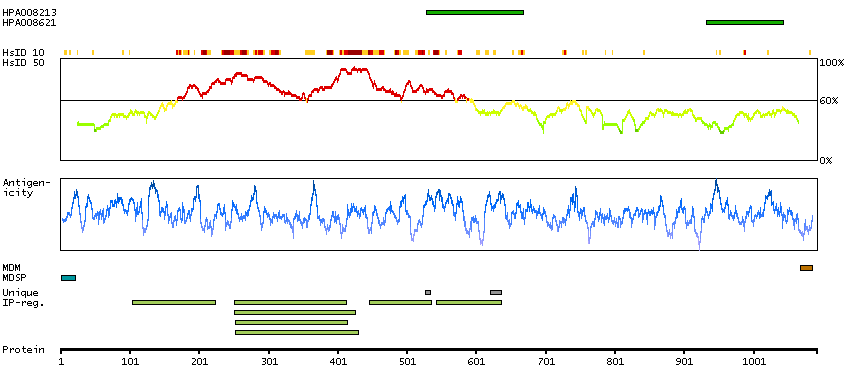
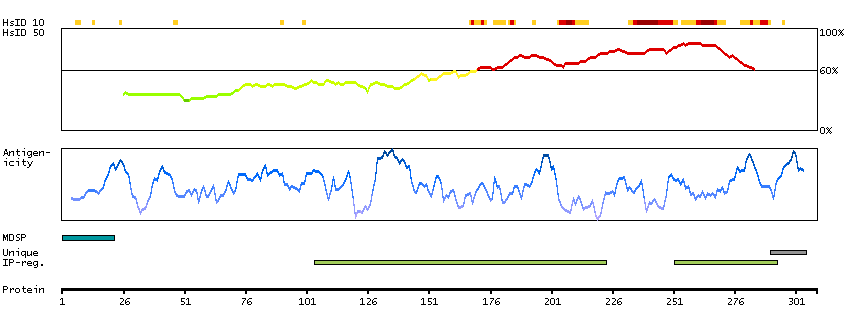
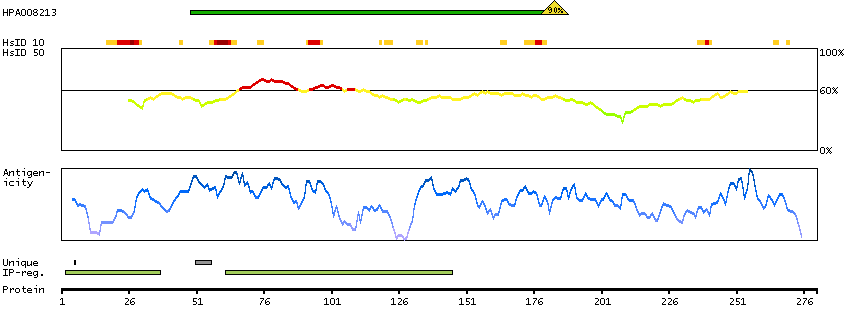
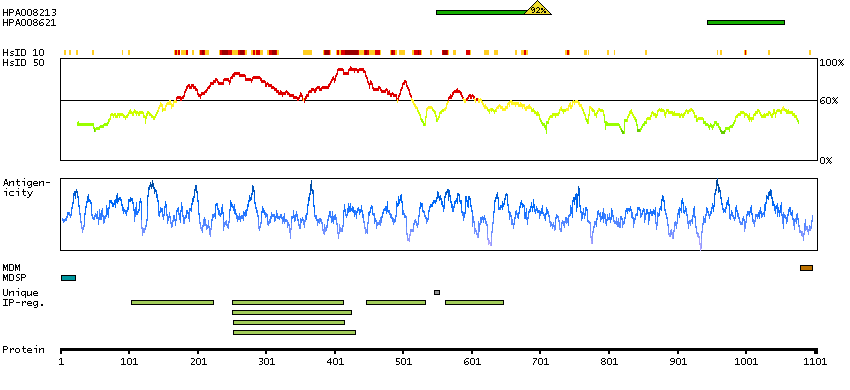
The preproprotein encoded by this gene is cleaved into multiple chains that comprise the alpha-2 and delta subunits of the voltage-dependent calcium channel complex. Calcium channels mediate the influx of calcium ions into the cell upon membrane polarization. Mutations in this gene can cause cardiac deficiencies, including Brugada syndrome and short QT syndrome. Alternate splicing results in multiple transcript variants, some of which may lack the delta subunit portion. [provided by RefSeq, Nov 2014]