We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
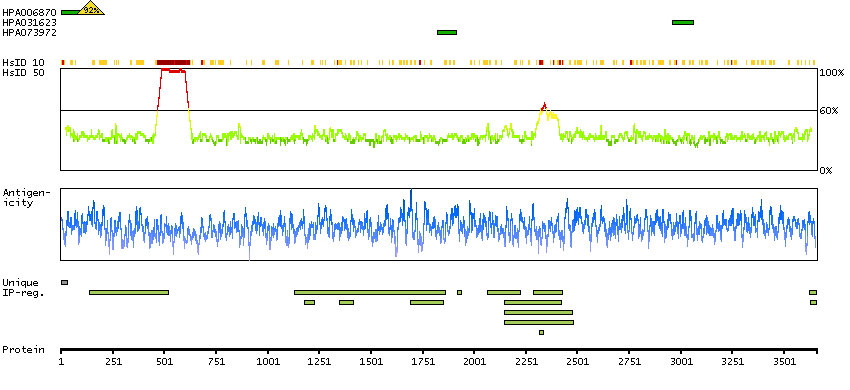
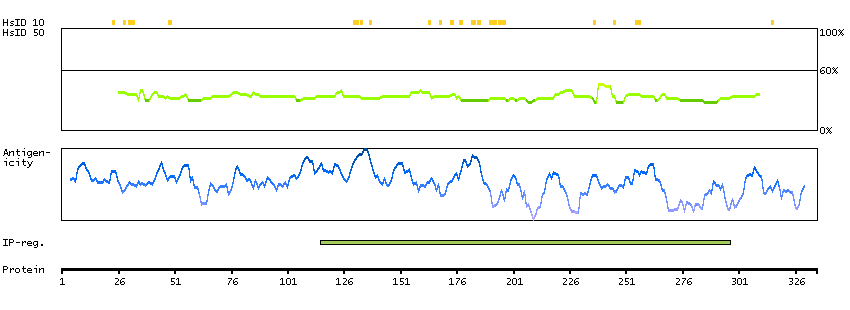
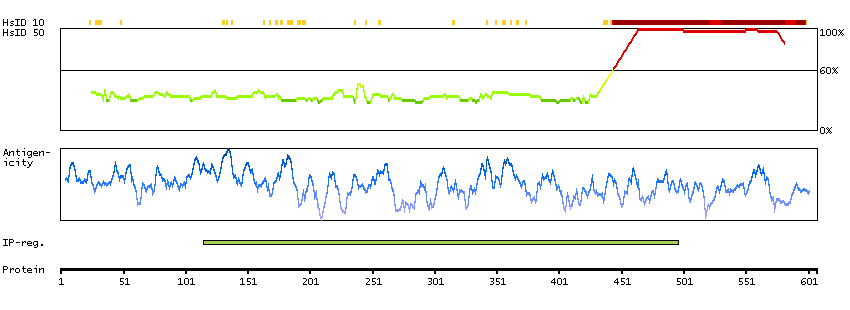
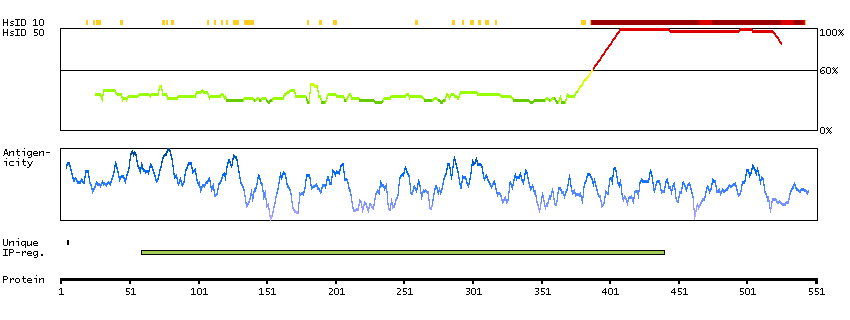
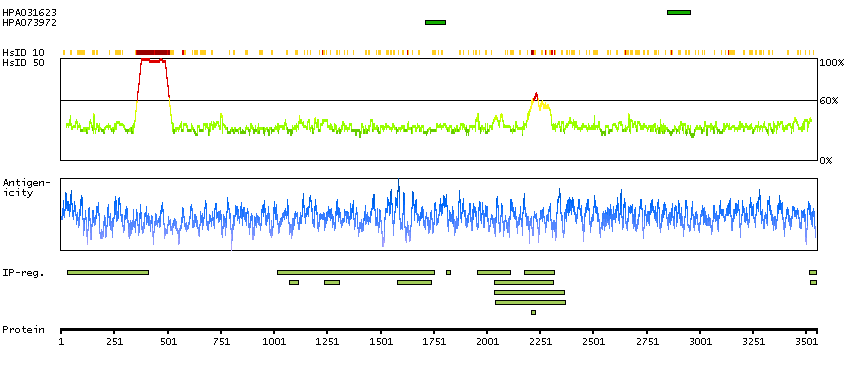
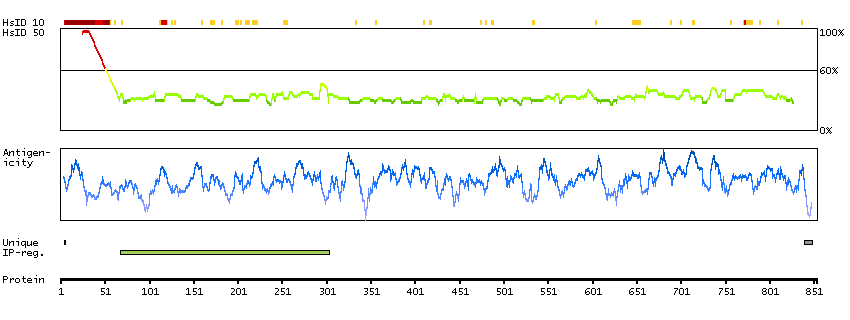
This gene encodes a protein involved in nonsense-mediated mRNA decay (NMD) as part of the mRNA surveillance complex. The protein has kinase activity and is thought to function in NMD by phosphorylating the regulator of nonsense transcripts 1 protein. Alternatively spliced transcript variants have been described, but their full-length nature has yet to be determined. [provided by RefSeq, Mar 2013]
Enzymes ENZYME proteins Transferases Kinases Atypical kinases SCAMPI predicted membrane proteins THUMBUP predicted membrane proteins Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)