We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
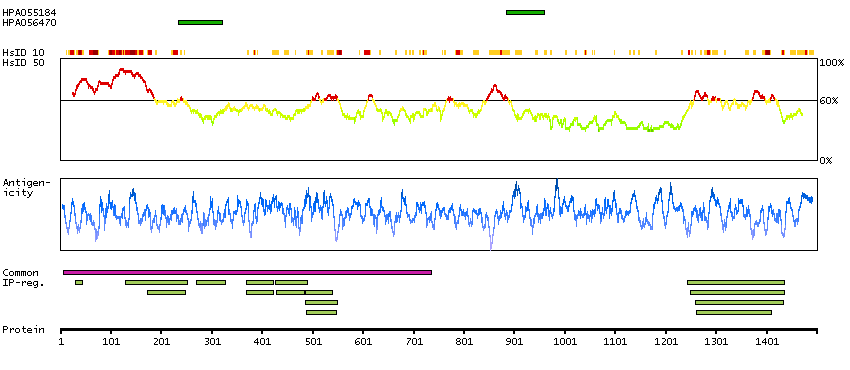
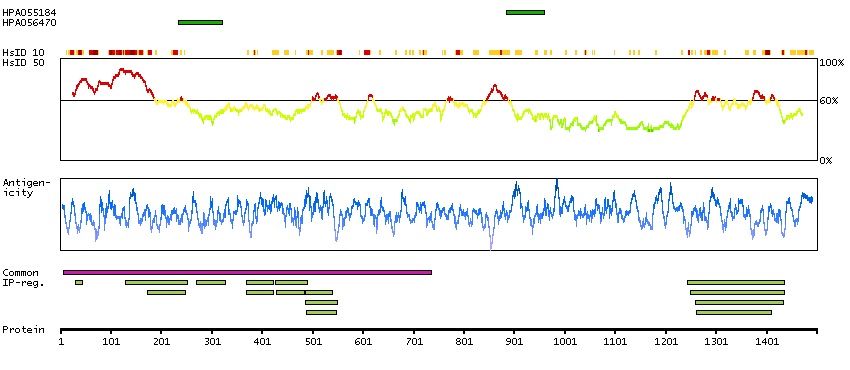
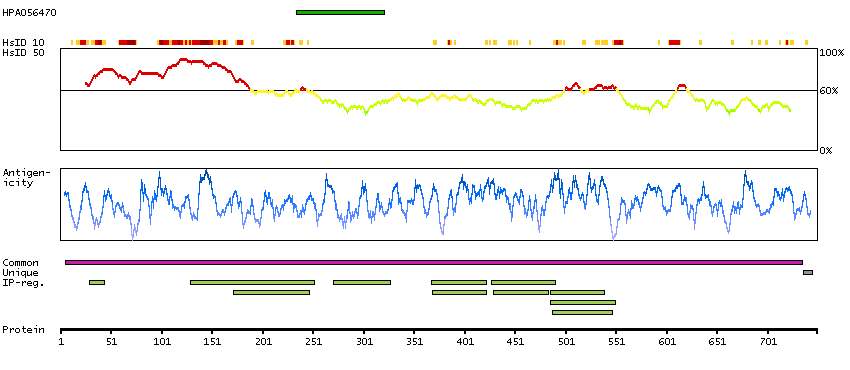
The human glucocorticoid receptor DNA binding factor, which associates with the promoter region of the glucocorticoid receptor gene (hGR gene), is a repressor of glucocorticoid receptor transcription. The amino acid sequence deduced from the cDNA sequences show the presence of three sequence motifs characteristic of a zinc finger and one motif suggestive of a leucine zipper in which 1 cysteine is found instead of all leucines. The GRLF1 enhances the homologous down-regulation of wild-type hGR gene expression. Biochemical analysis suggests that GRLF1 interaction is sequence specific and that transcriptional efficacy of GRLF1 is regulated through its interaction with specific sequence motif. The level of expression is regulated by glucocorticoids. [provided by RefSeq, Jul 2008]
Q9NRY4 [Direct mapping] Rho GTPase-activating protein 35
Show all
Phobius predicted membrane proteins Predicted intracellular proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0001227 [transcriptional repressor activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0001843 [neural tube closure] GO:0003677 [DNA binding] GO:0003714 [transcription corepressor activity] GO:0005096 [GTPase activator activity] GO:0005525 [GTP binding] GO:0005634 [nucleus] GO:0005737 [cytoplasm] GO:0005829 [cytosol] GO:0006351 [transcription, DNA-templated] GO:0007165 [signal transduction] GO:0007264 [small GTPase mediated signal transduction] GO:0007411 [axon guidance] GO:0008360 [regulation of cell shape] GO:0010976 [positive regulation of neuron projection development] GO:0015629 [actin cytoskeleton] GO:0030900 [forebrain development] GO:0035024 [negative regulation of Rho protein signal transduction] GO:0043010 [camera-type eye development] GO:0043116 [negative regulation of vascular permeability] GO:0043547 [positive regulation of GTPase activity] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0051056 [regulation of small GTPase mediated signal transduction]
Q9NRY4 [Direct mapping] Rho GTPase-activating protein 35
Show all
Phobius predicted membrane proteins Predicted intracellular proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0001227 [transcriptional repressor activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0003677 [DNA binding] GO:0003714 [transcription corepressor activity] GO:0005096 [GTPase activator activity] GO:0005525 [GTP binding] GO:0005634 [nucleus] GO:0005829 [cytosol] GO:0006351 [transcription, DNA-templated] GO:0007165 [signal transduction] GO:0007264 [small GTPase mediated signal transduction] GO:0007411 [axon guidance] GO:0043547 [positive regulation of GTPase activity] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0051056 [regulation of small GTPase mediated signal transduction]
A2RRE5 [Direct mapping] GRLF1 protein; Rho GTPase-activating protein 35
Show all
Phobius predicted membrane proteins Predicted intracellular proteins Cancer-related genes Mutated cancer genes Mutational cancer driver genes Protein evidence (Ezkurdia et al 2014)