We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein encoded by this gene acts as a homotetramer to catalyze the conversion of homocysteine to cystathionine, the first step in the transsulfuration pathway. The encoded protein is allosterically activated by adenosyl-methionine and uses pyridoxal phosphate as a cofactor. Defects in this gene can cause cystathionine beta-synthase deficiency (CBSD), which can lead to homocystinuria. Multiple alternatively spliced transcript variants have been found for this gene. [provided by RefSeq, May 2010]
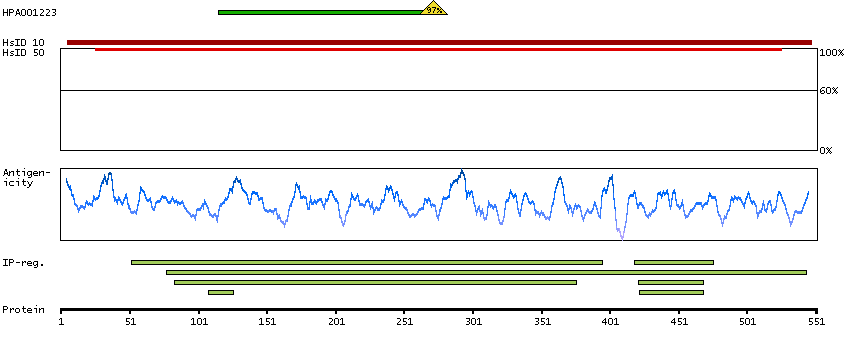
Enzymes ENZYME proteins Lyases Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
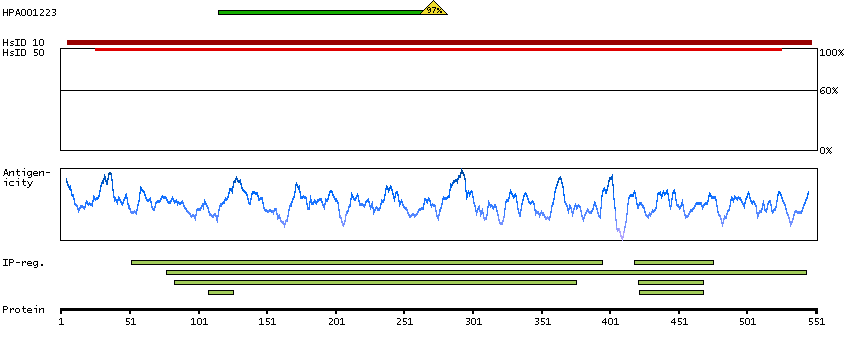
Enzymes ENZYME proteins Lyases Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
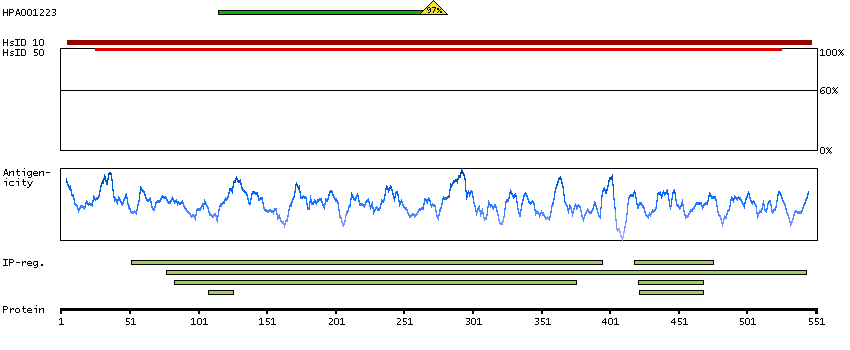
Enzymes ENZYME proteins Lyases Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
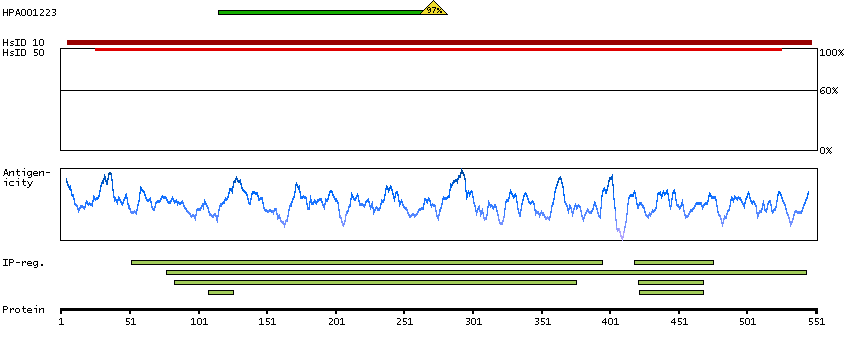
Enzymes ENZYME proteins Lyases Predicted intracellular proteins Plasma proteins Disease related genes Potential drug targets Protein evidence (Ezkurdia et al 2014)