We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
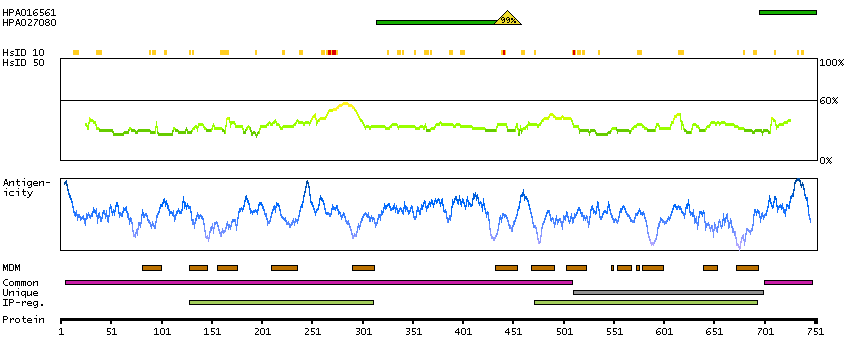
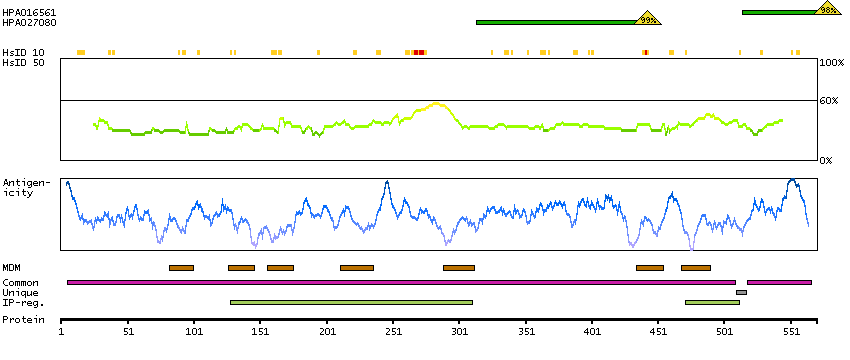
This gene encodes a putative cation-selective ion channel with two repeats of a six-transmembrane-domain. The protein localizes to lysosomal membranes and enables nicotinic acid adenine dinucleotide phosphate (NAADP) -induced calcium ion release from lysosome-related stores. This ubiquitously expressed gene has elevated expression in liver and kidney. Two common nonsynonymous SNPs in this gene strongly associate with blond versus brown hair pigmentation.[provided by RefSeq, Dec 2009]