We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
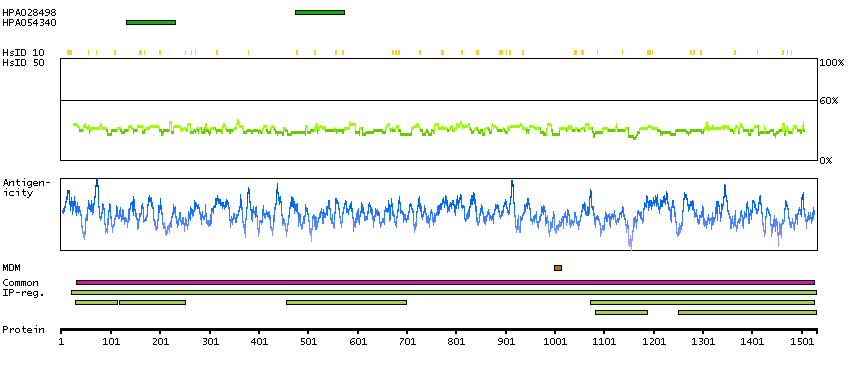
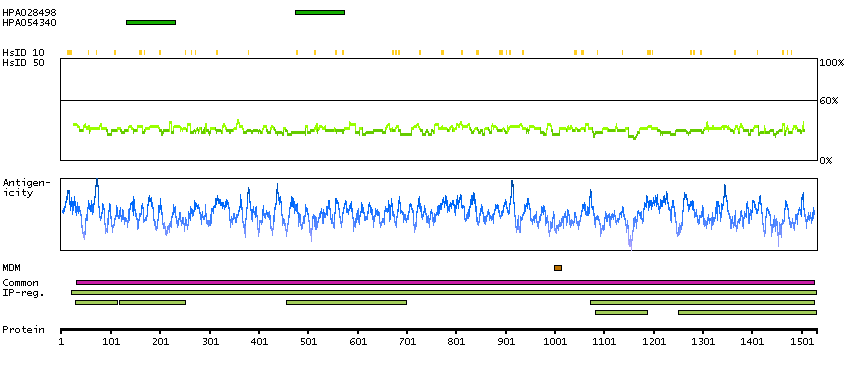
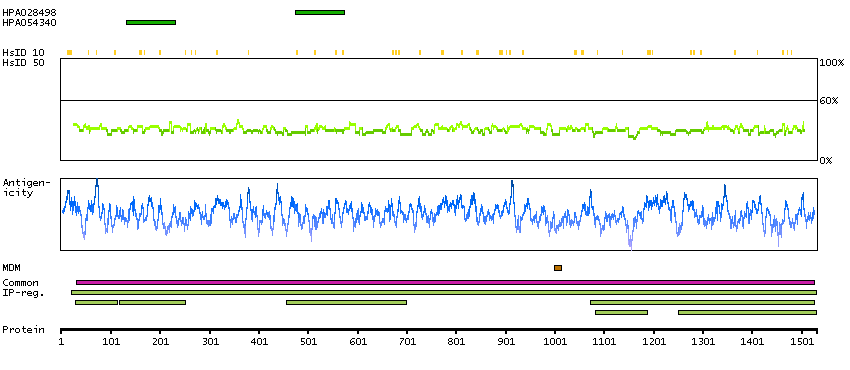
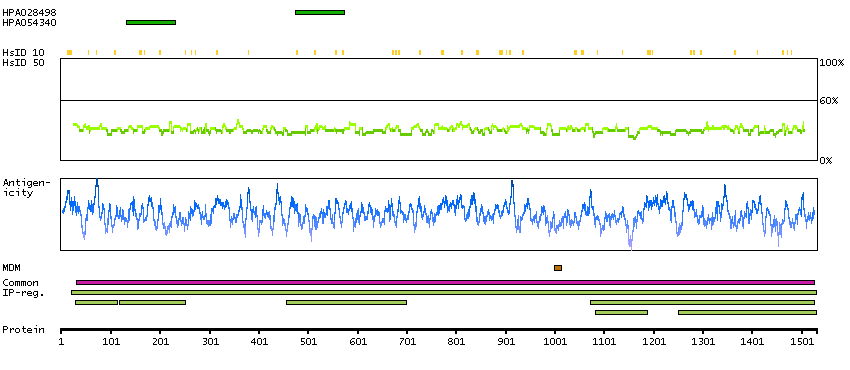
This gene encodes the glycogen debrancher enzyme which is involved in glycogen degradation. This enzyme has two independent catalytic activities which occur at different sites on the protein: a 4-alpha-glucotransferase activity and a amylo-1,6-glucosidase activity. Mutations in this gene are associated with glycogen storage disease although a wide range of enzymatic and clinical variability occurs which may be due to tissue-specific alternative splicing. Alternatively spliced transcripts encoding different isoforms have been described. [provided by RefSeq, Jul 2008]