We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
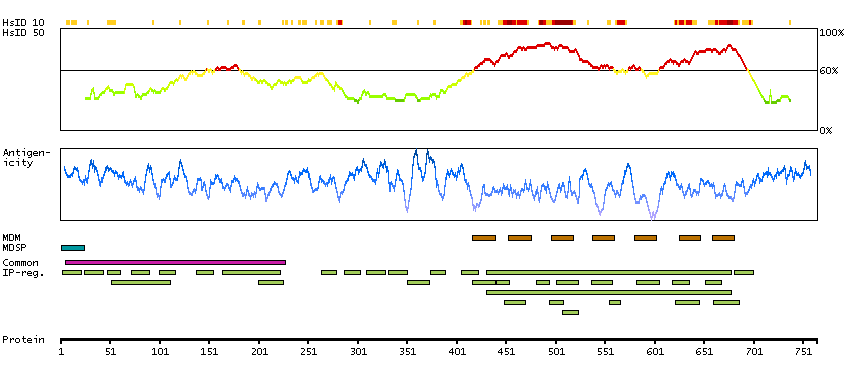
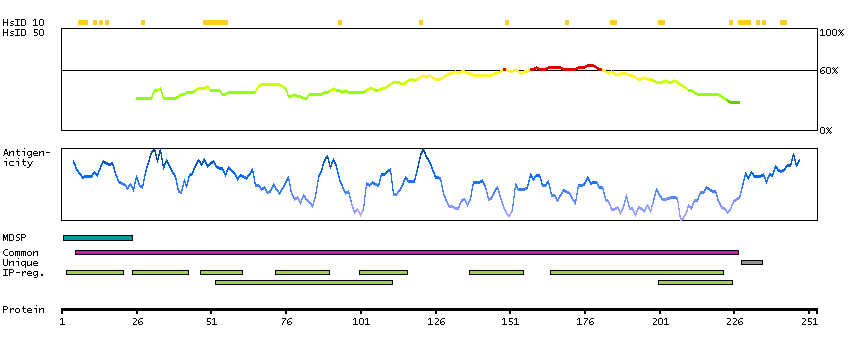
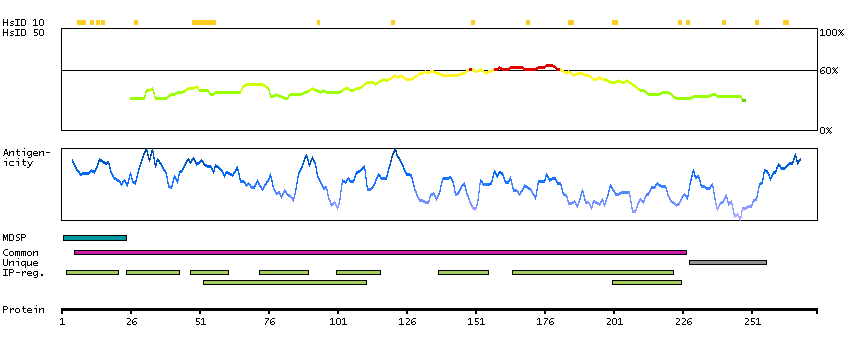
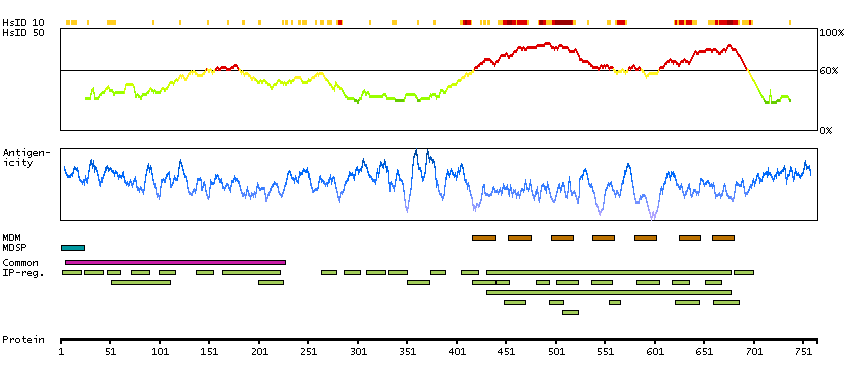
The protein encoded by this gene is a membrane protein and a major controller of thyroid cell metabolism. The encoded protein is a receptor for thyrothropin and thyrostimulin, and its activity is mediated by adenylate cyclase. Defects in this gene are a cause of several types of hyperthyroidism. Three transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Dec 2008]