We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
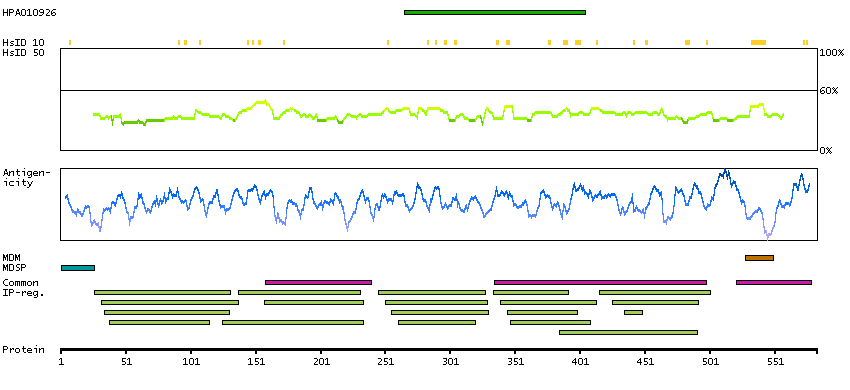
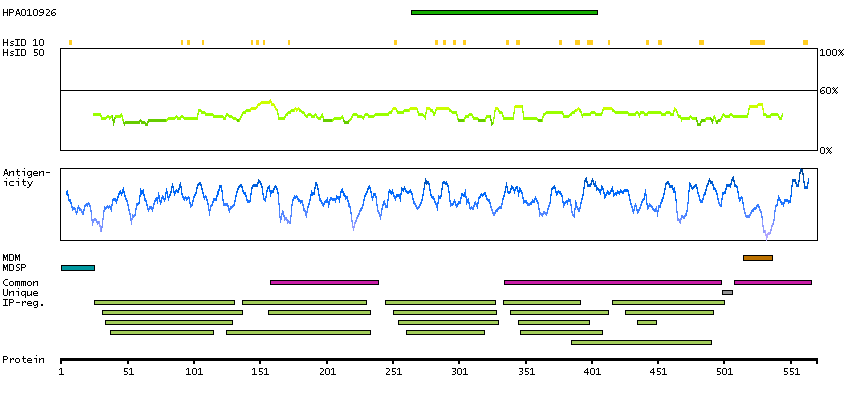
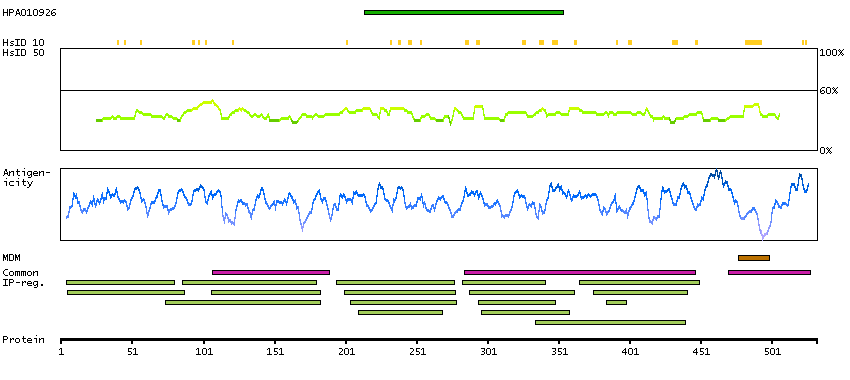
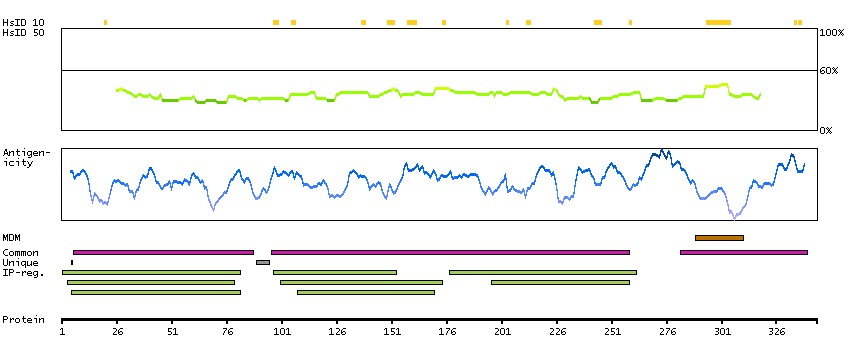
This gene encodes activated leukocyte cell adhesion molecule (ALCAM), also known as CD166 (cluster of differentiation 166), which is a member of a subfamily of immunoglobulin receptors with five immunoglobulin-like domains (VVC2C2C2) in the extracellular domain. This protein binds to T-cell differentiation antigene CD6, and is implicated in the processes of cell adhesion and migration. Multiple alternatively spliced transcript variants encoding different isoforms have been found. [provided by RefSeq, Aug 2011]