We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
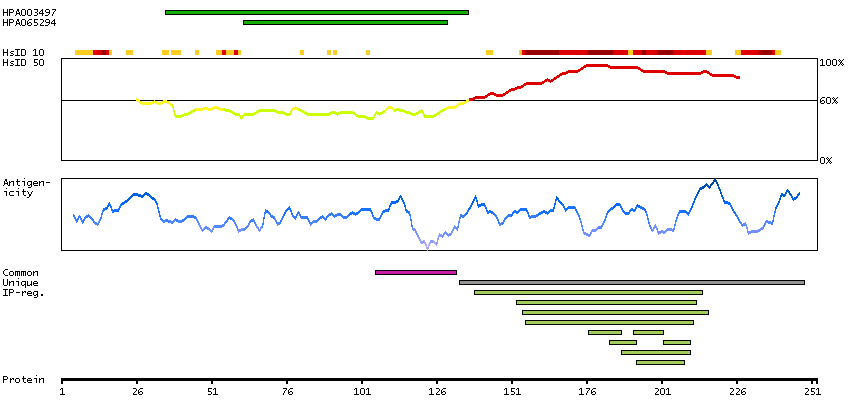
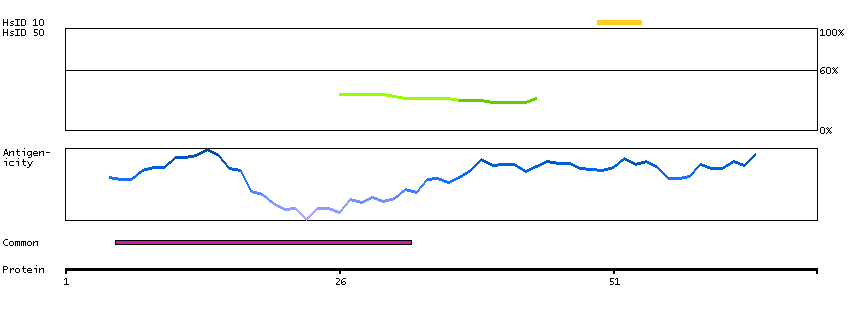
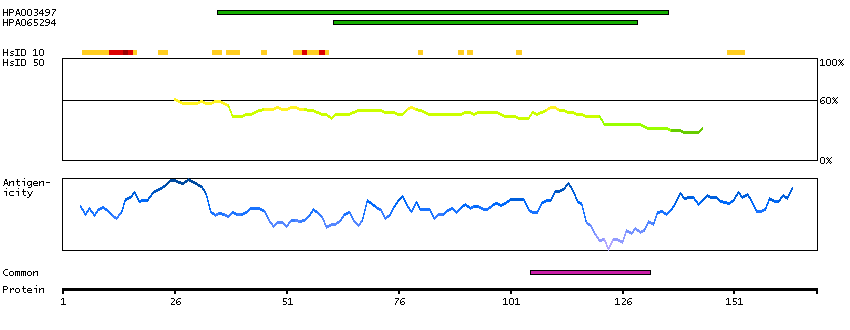
This gene encodes a homeobox-containing transcription factor that is the homolog to the 'empty spiracles' gene in Drosophila. Research on this gene in humans has focused on its expression in three tissues: dorsal telencephalon, olfactory neuroepithelium, and urogenetial system. It is expressed in the dorsal telencephalon during development in a low rostral-lateral to high caudal-medial gradient and is proposed to pattern the neocortex into defined functional areas. It is also expressed in embryonic and adult olfactory neuroepithelia where it complexes with eukaryotic translation initiation factor 4E (eIF4E) and possibly regulates mRNA transport or translation. In the developing urogenital system, it is expressed in epithelial tissues and is negatively regulated by HOXA10. Alternative splicing results in multiple transcript variants encoding distinct proteins.[provided by RefSeq, Sep 2009]