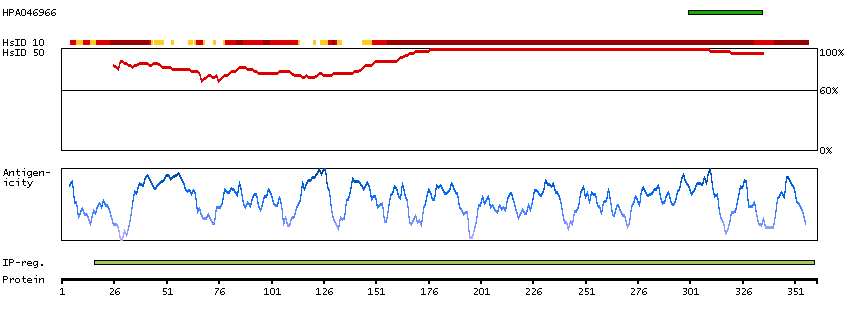
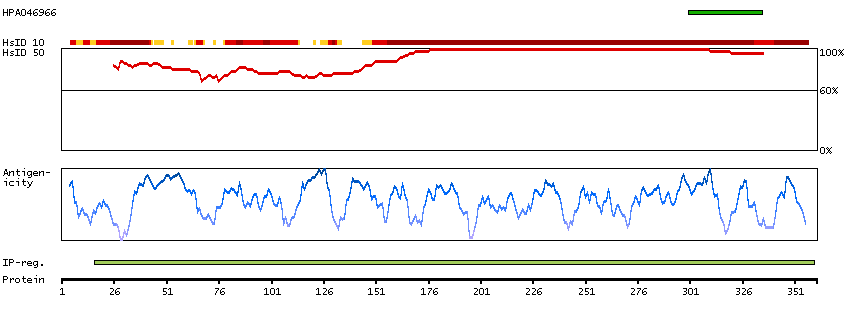
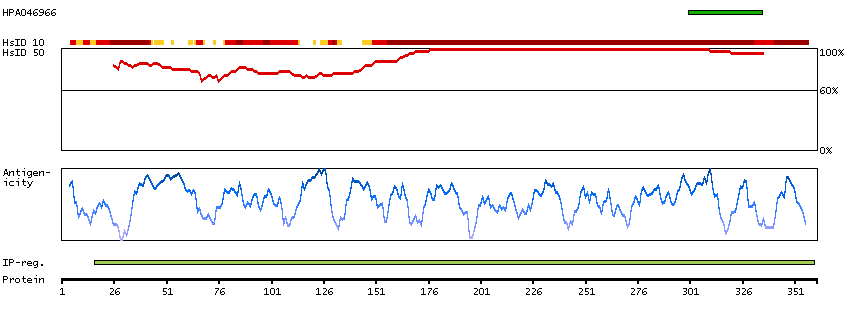
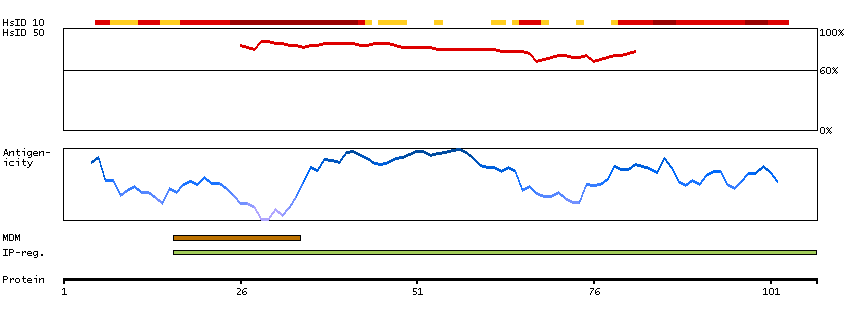
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Fucosyltransferase 3 (galactoside 3(4)-L-fucosyltransferase, Lewis blood group) (HGNC Symbol)
Entrez gene summary
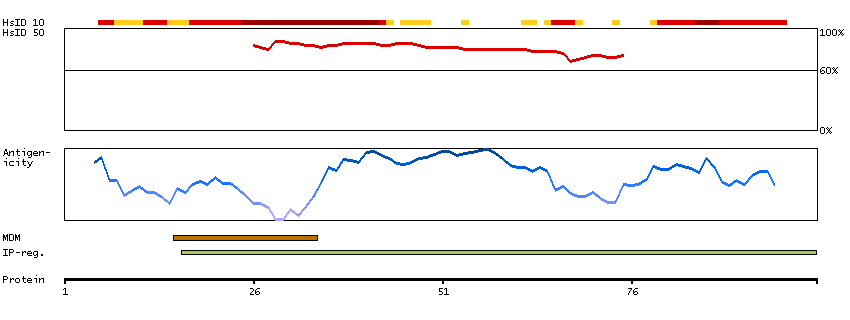
The Lewis histo-blood group system comprises a set of fucosylated glycosphingolipids that are synthesized by exocrine epithelial cells and circulate in body fluids. The glycosphingolipids function in embryogenesis, tissue differentiation, tumor metastasis, inflammation, and bacterial adhesion. They are secondarily absorbed to red blood cells giving rise to their Lewis phenotype. This gene is a member of the fucosyltransferase family, which catalyzes the addition of fucose to precursor polysaccharides in the last step of Lewis antigen biosynthesis. It encodes an enzyme with alpha(1,3)-fucosyltransferase and alpha(1,4)-fucosyltransferase activities. Mutations in this gene are responsible for the majority of Lewis antigen-negative phenotypes. Multiple alternatively spliced variants, encoding the same protein, have been found for this gene. [provided by RefSeq, Jul 2008]