We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
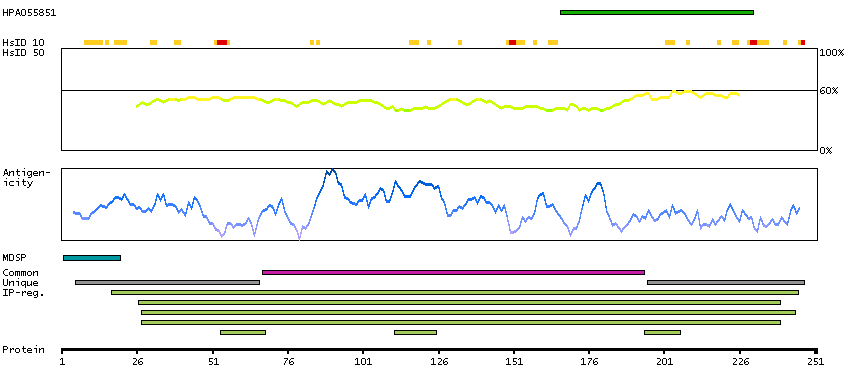
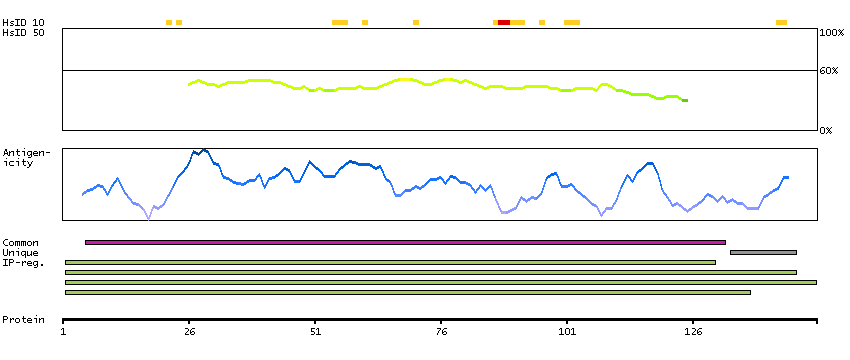
Azurophil granules, specialized lysosomes of the neutrophil, contain at least 10 proteins implicated in the killing of microorganisms. This gene encodes a preproprotein that is proteolytically processed to generate a mature azurophil granule antibiotic protein, with monocyte chemotactic and antimicrobial activity. It is also an important multifunctional inflammatory mediator. This encoded protein is a member of the serine protease gene family but it is not a serine proteinase, because the active site serine and histidine residues are replaced. The genes encoding this protein, neutrophil elastase 2, and proteinase 3 are in a cluster located at chromosome 19pter. All 3 genes are expressed coordinately and their protein products are packaged together into azurophil granules during neutrophil differentiation. [provided by RefSeq, Nov 2015]