We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Group enriched (epididymis, thyroid gland)
GENE INFORMATION
Gene name
RAG2
Synonyms
Description
Recombination activating gene 2 (HGNC Symbol)
Entrez gene summary
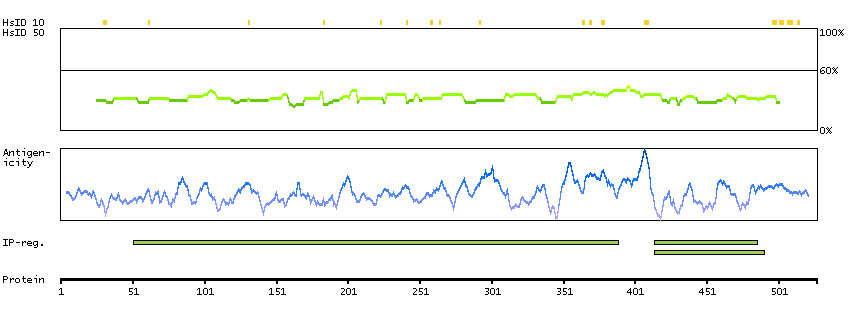
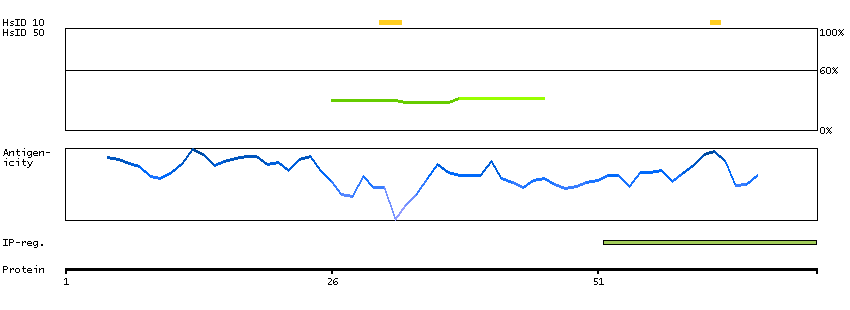
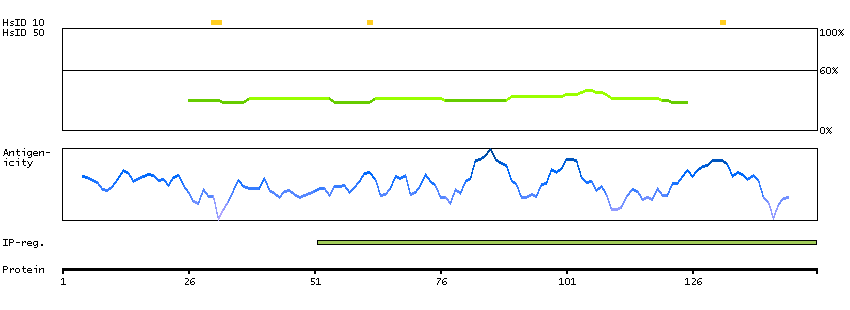
This gene encodes a protein that is involved in the initiation of V(D)J recombination during B and T cell development. This protein forms a complex with the product of the adjacent recombination activating gene 1, and this complex can form double-strand breaks by cleaving DNA at conserved recombination signal sequences. The recombination activating gene 1 component is thought to contain most of the catalytic activity, while the N-terminal of the recombination activating gene 2 component is thought to form a six-bladed propeller in the active core that serves as a binding scaffold for the tight association of the complex with DNA. A C-terminal plant homeodomain finger-like motif in this protein is necessary for interactions with chromatin components, specifically with histone H3 that is trimethylated at lysine 4. Mutations in this gene cause Omenn syndrome, a form of severe combined immunodeficiency associated with autoimmune-like symptoms. [provided by RefSeq, Jul 2008]