We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
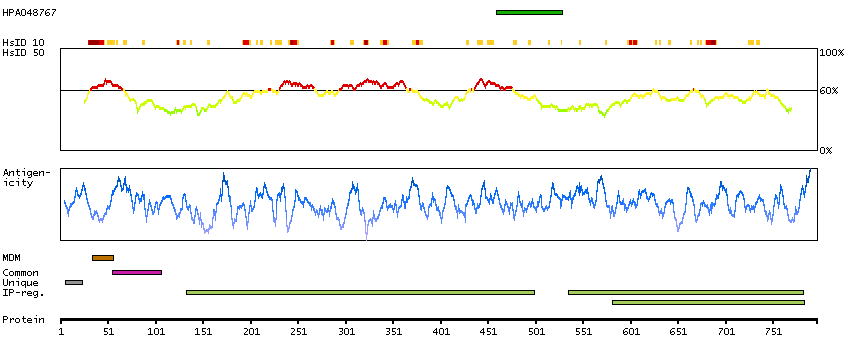
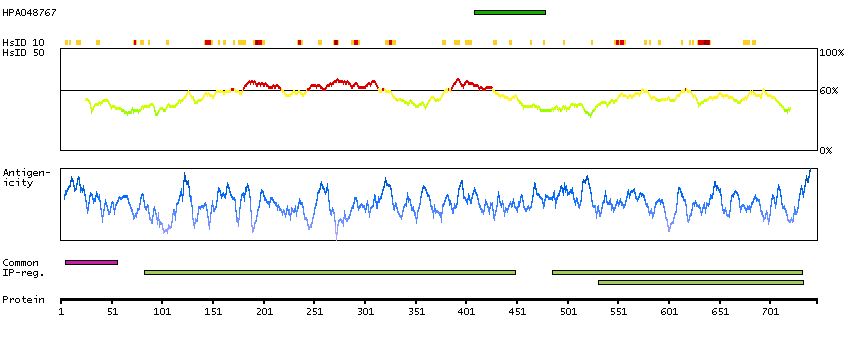
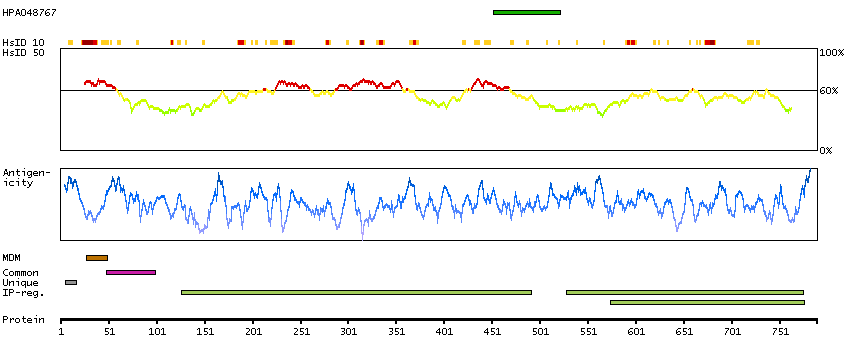
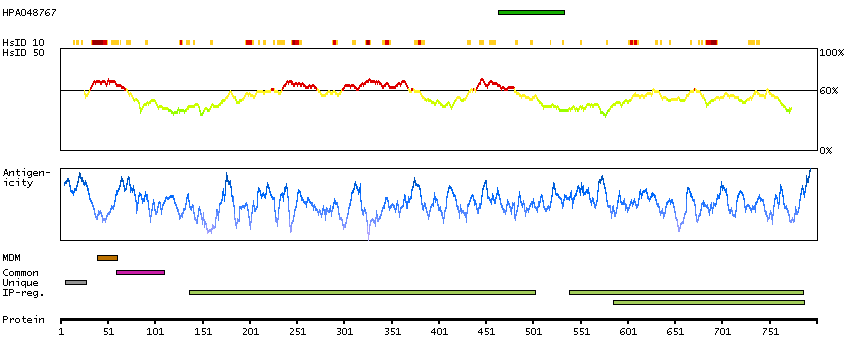
This gene encodes a single-pass type II membrane protein that is a member of the S9B family in clan SC of the serine proteases. This protein has no detectable protease activity, most likely due to the absence of the conserved serine residue normally present in the catalytic domain of serine proteases. However, it does bind specific voltage-gated potassium channels and alters their expression and biophysical properties. Mutations in this gene have been associated with asthma. Alternate transcriptional splice variants, encoding different isoforms, have been characterized. [provided by RefSeq, Jul 2008]
Enzymes Peptidases Serine-type peptidases Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0005886 [plasma membrane] GO:0006508 [proteolysis] GO:0008236 [serine-type peptidase activity] GO:0015459 [potassium channel regulator activity] GO:0016020 [membrane] GO:0016021 [integral component of membrane] GO:0072659 [protein localization to plasma membrane] GO:0090004 [positive regulation of establishment of protein localization to plasma membrane] GO:1901379 [regulation of potassium ion transmembrane transport]