We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
RNA tissue category: Tissue enhanced (salivary gland, small intestine)
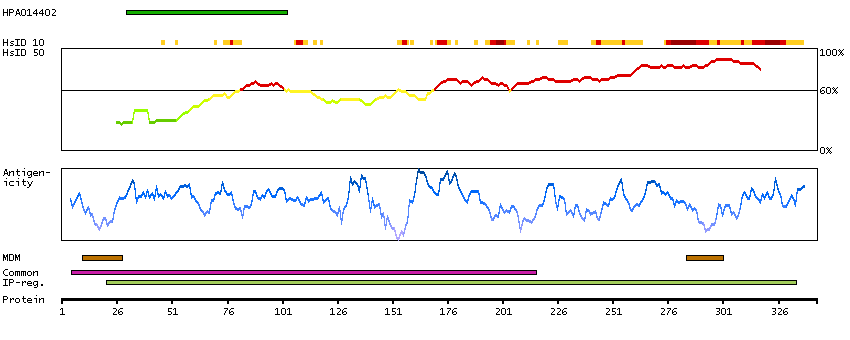
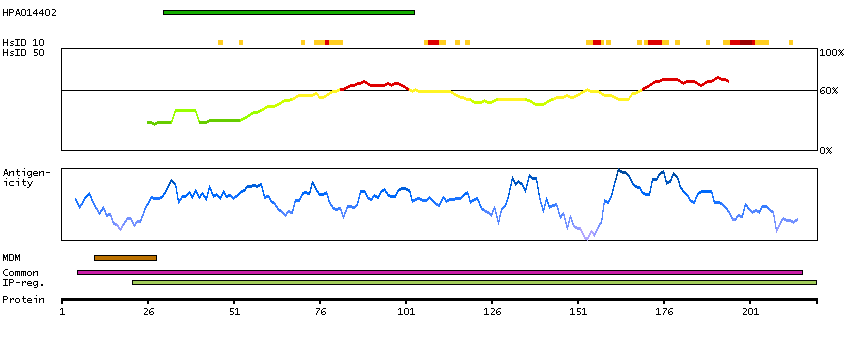
GENE INFORMATION
Gene name
FUT2 (HGNC Symbol)
Synonyms
SE, Se2, SEC2, sej
Description
Fucosyltransferase 2 (secretor status included) (HGNC Symbol)
Entrez gene summary
The protein encoded by this gene is a Golgi stack membrane protein that is involved in the creation of a precursor of the H antigen, which is required for the final step in the soluble A and B antigen synthesis pathway. This gene is one of two encoding the galactoside 2-L-fucosyltransferase enzyme. Two transcript variants encoding the same protein have been found for this gene. [provided by RefSeq, Jul 2008]