We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
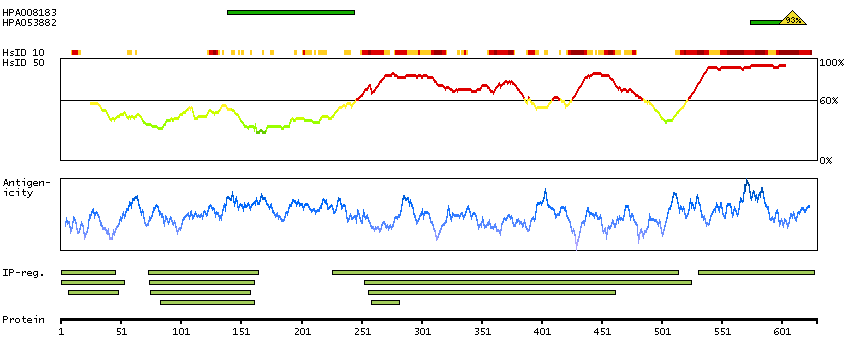
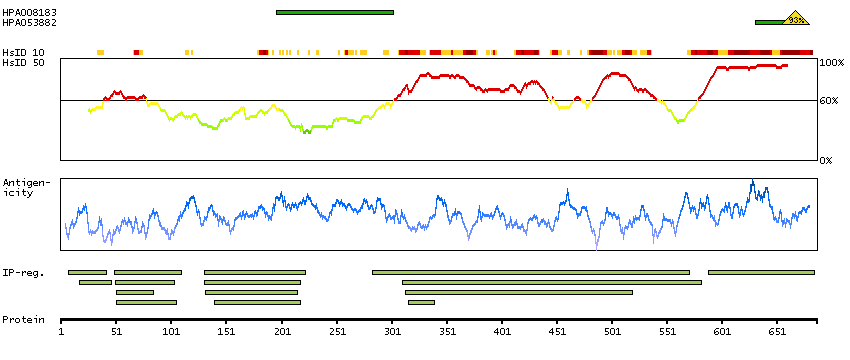
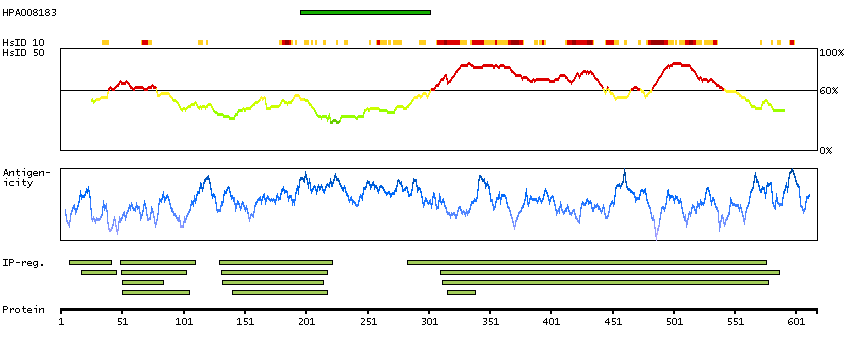
There are approximately 40 known eukaryotic LIM proteins, so named for the LIM domains they contain. LIM domains are highly conserved cysteine-rich structures containing 2 zinc fingers. Although zinc fingers usually function by binding to DNA or RNA, the LIM motif probably mediates protein-protein interactions. LIM kinase-1 and LIM kinase-2 belong to a small subfamily with a unique combination of 2 N-terminal LIM motifs and a C-terminal protein kinase domain. The protein encoded by this gene is phosphorylated and activated by ROCK, a downstream effector of Rho, and the encoded protein, in turn, phosphorylates cofilin, inhibiting its actin-depolymerizing activity. It is thought that this pathway contributes to Rho-induced reorganization of the actin cytoskeleton. At least three transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Transferases Kinases TKL Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Transferases Kinases TKL Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Enzymes ENZYME proteins Transferases Kinases TKL Ser/Thr protein kinases Predicted intracellular proteins Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)