We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
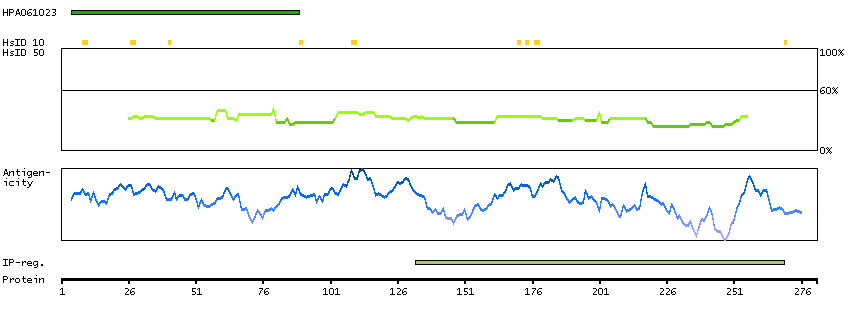
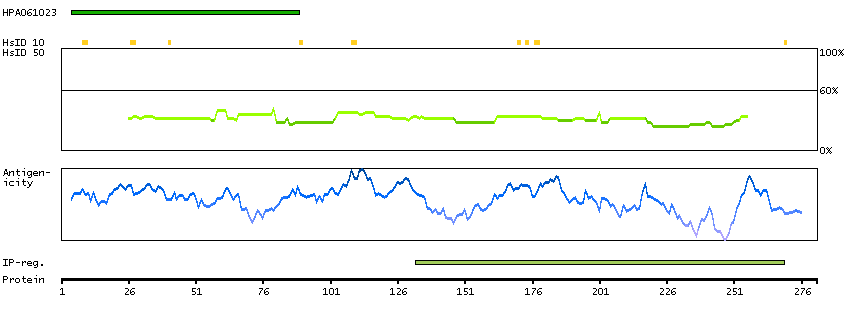
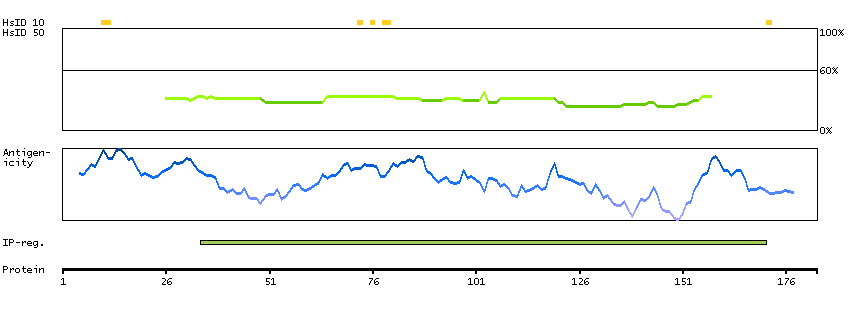
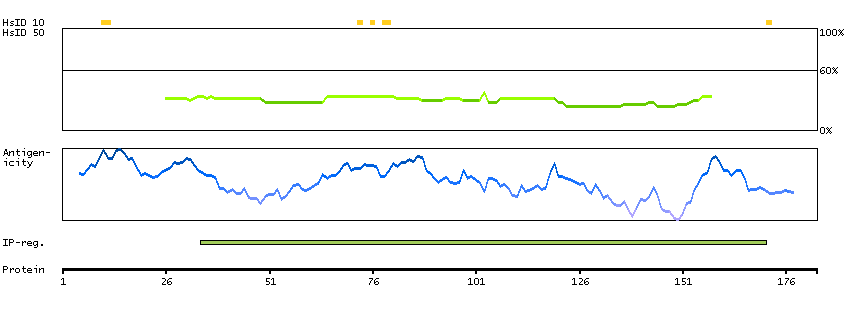
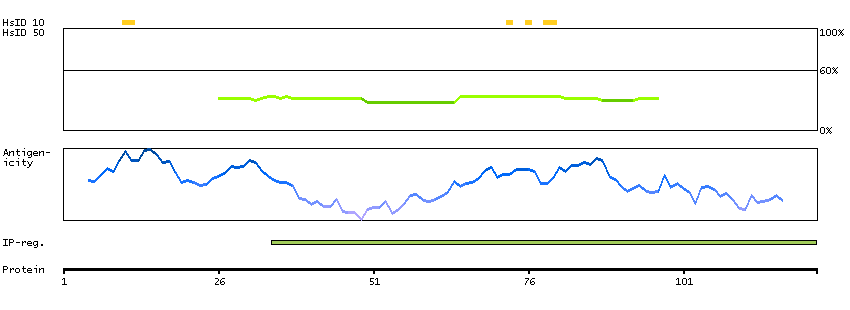
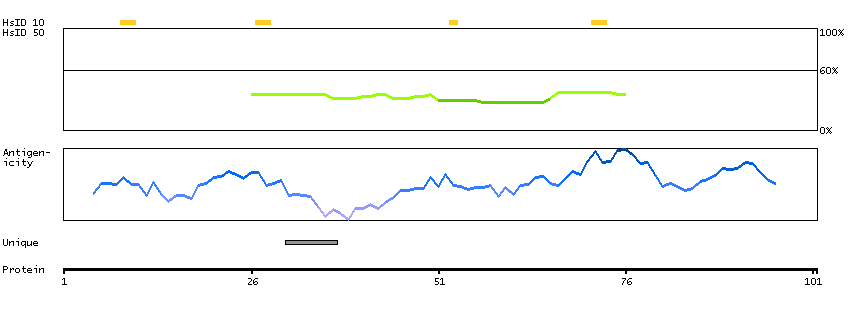
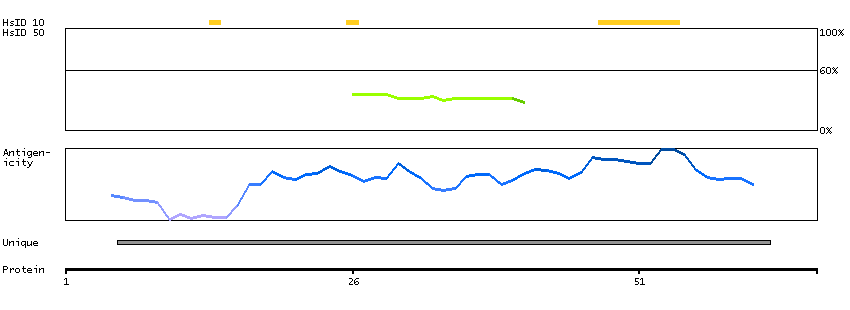
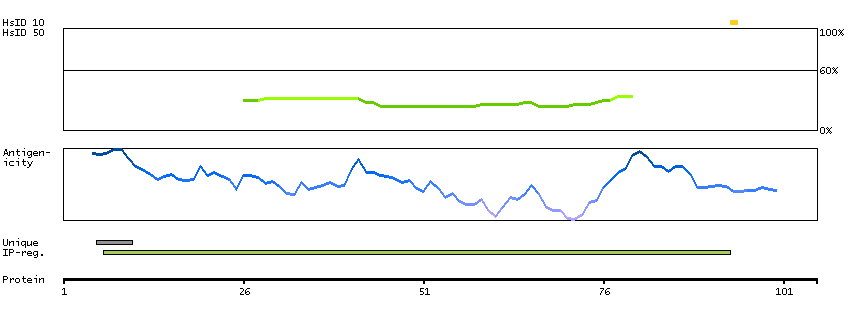
This gene encodes a protein with two predicted helix-turn-helix domains. Mutations in this gene were found in families with congenital dyserythropoietic anemia type Ib. Alternative splicing results in multiple transcript variants encoding different isoforms. [provided by RefSeq, Mar 2014]