We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
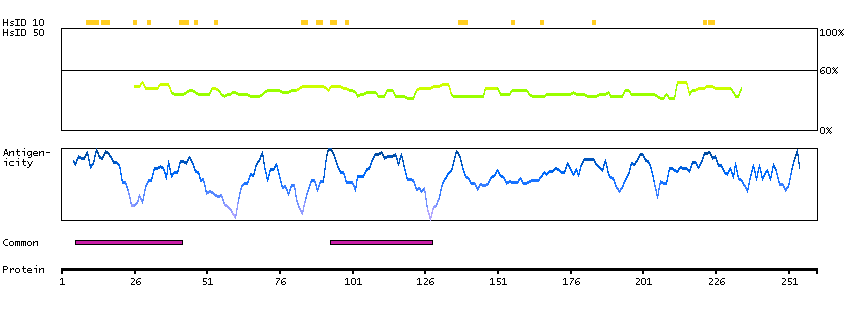
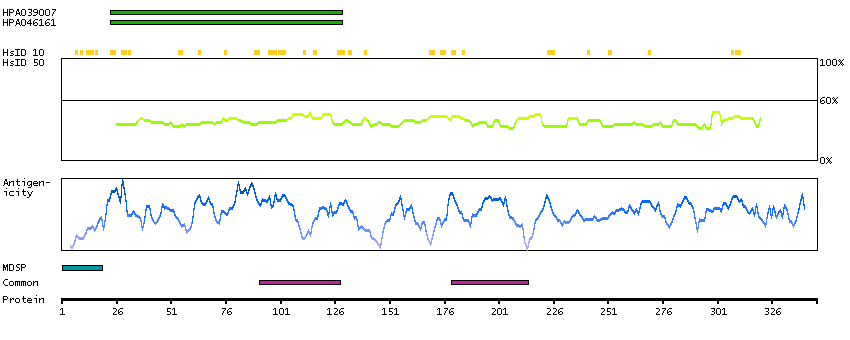
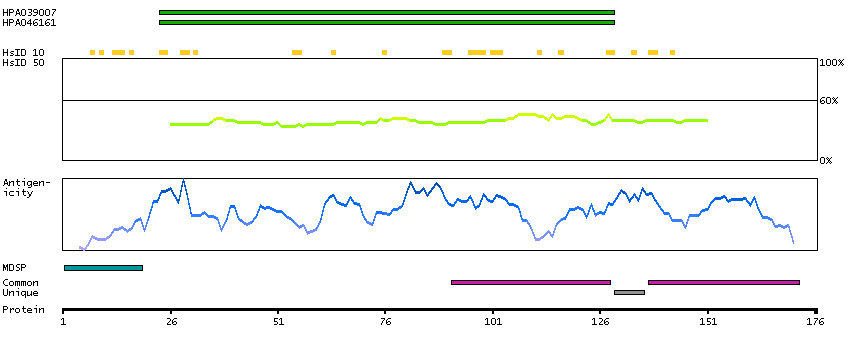
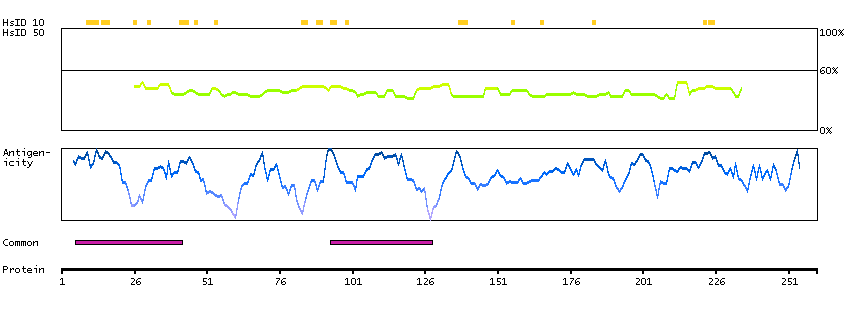
This gene encodes a leucine zipper protein. This protein is deleted in some patients with Wilms tumor-Aniridia-Genitourinary anomalies-mental Retardation (WAGR) syndrome. Alternate splicing results in multiple transcript variants. [provided by RefSeq, Oct 2011]