We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
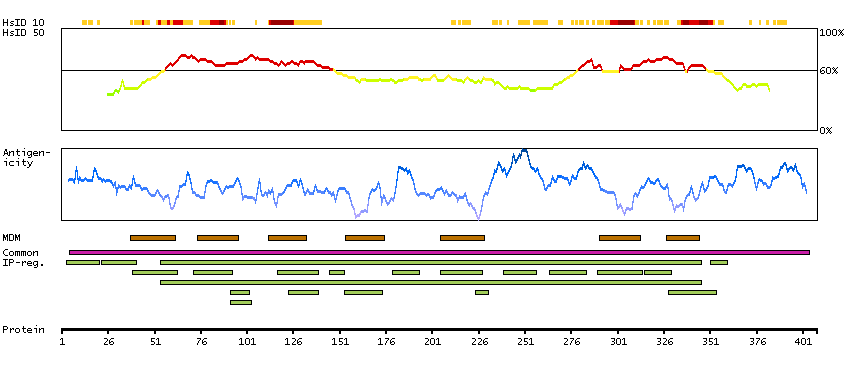
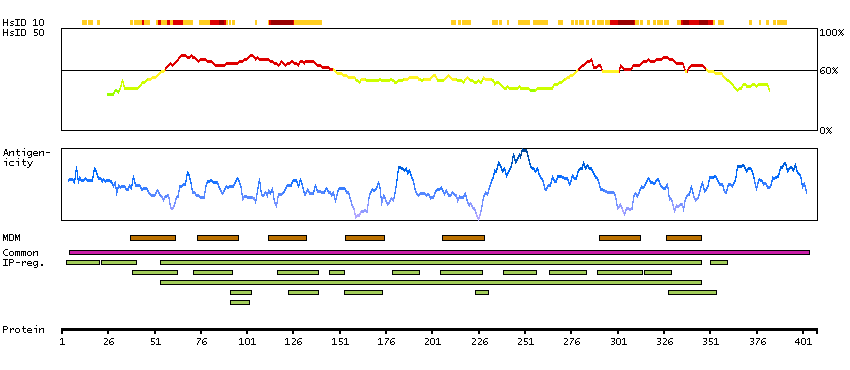
The protein encoded by this gene belongs to the family of beta adrenergic receptors, which mediate catecholamine-induced activation of adenylate cyclase through the action of G proteins. This receptor is located mainly in the adipose tissue and is involved in the regulation of lipolysis and thermogenesis. [provided by RefSeq, Feb 2009]